CONFLEX計算の詳細設定
計算設定の初期ダイアログの、「Detail Settings」ボタンをクリックすると、詳細設定ダイアログが表示されます。ここでは、より細かい計算設定が可能です。ダイアログは、各設定ダイアログの埋め込み形式になっており、全体を見渡すことが可能です。
ダイアログ左下の「Basic Settings」ボタンをクリックすると、元の簡易設定ダイアログに戻ります。
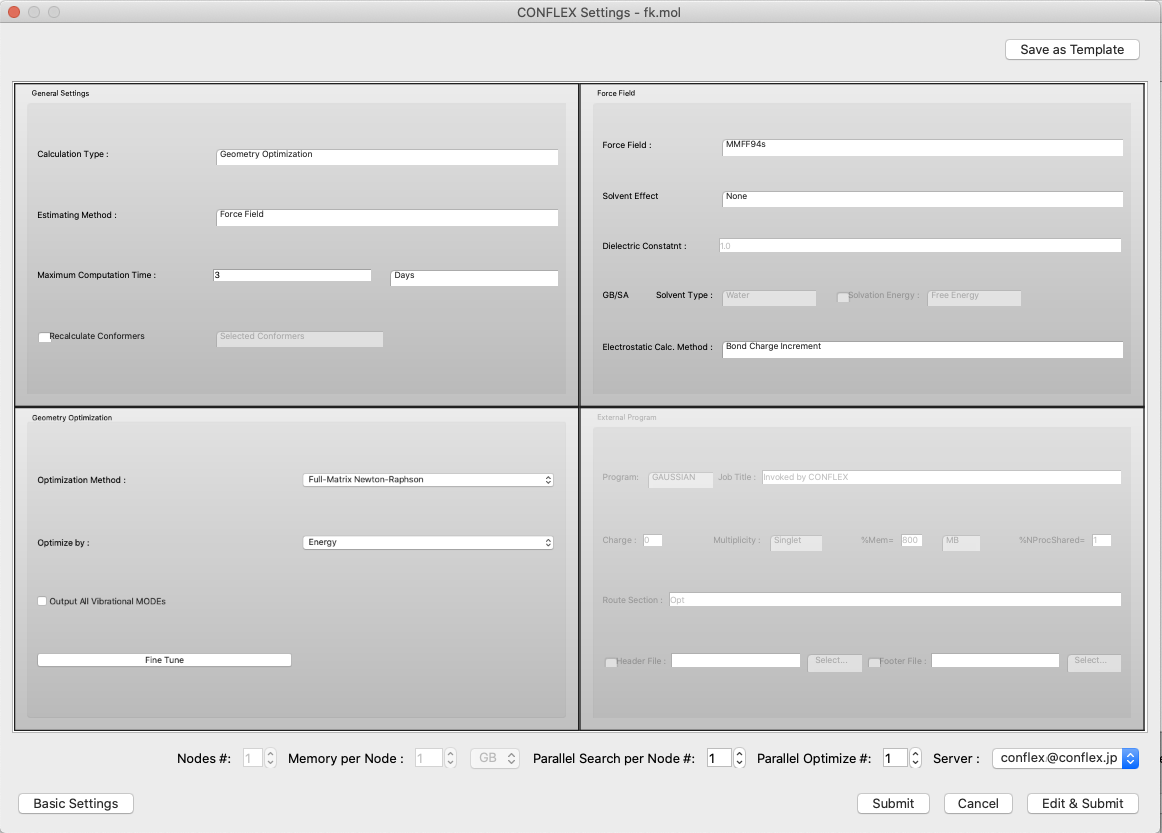
- Parallel Search #およびParallel Optimize #
-
並列版CONFLEXをお持ちの方は、「Parallel Search #」および「Parallel Optimize #」で1より大きな数値を設定することで、並列計算が実行されます。
前者は、配座探索時の各構造を個別のCPUで最適化するため、配座探索計算が高速化されます。
後者は、構造の最適化計算を複数のCPUを用いて行って高速化します。
通常の分子でしたら、「Parallel Search」だけ設定した方が効率がいいでしょう。結晶などのように構造が大きな場合、両者を併用して構造最適化も高速化すれば、全体が高速化されます。
- Save as Template
-
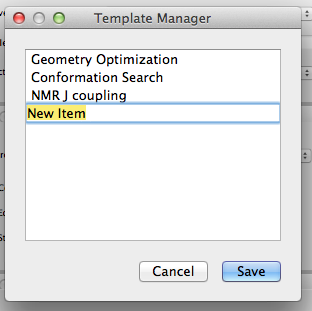
ダイアログ右上の「Save as Template」ボタンをクリックすると、現在の設定をテンプレートとして保存できます。以下のようなダイアログが現れ ますので、「New Item」を任意の文字列に変更してください。これらは、簡易設定ダイアログから選択できるようになります。
また、これらの設定内容は、Applicationsメニューから「Scheme Editor」を選択し編集プログラムを起動して編集する事が可能です。
- Server:
- 計算を行う場所を「Server :」で指定します。
- Submit
- ジョブは「Submit」ボタンをクリックすると、投入されます。
- Edit & Submit
- 手作業で設定を書き換えるには、「Edit & Submit」ボタンをクリックしてテキストエディターを開きます。ダイアログ項目に無い設定を行いたい場合に利用します。
- CONFLEX計算設定ウィンドウのメインエリアに埋め込まれているダイアログボックスには、以下のものが有ります:
-
- General Settings
- Force Field
- Geometry Optimization
- Conformation Search
- Solvent Effect
- UV/Vis/CD Spectrum
- Crystal Optimization
- Crystal Search
- Host Ligand Search
- Dynamic Reaction Coordinate
- External Program
General Settingsダイアログ
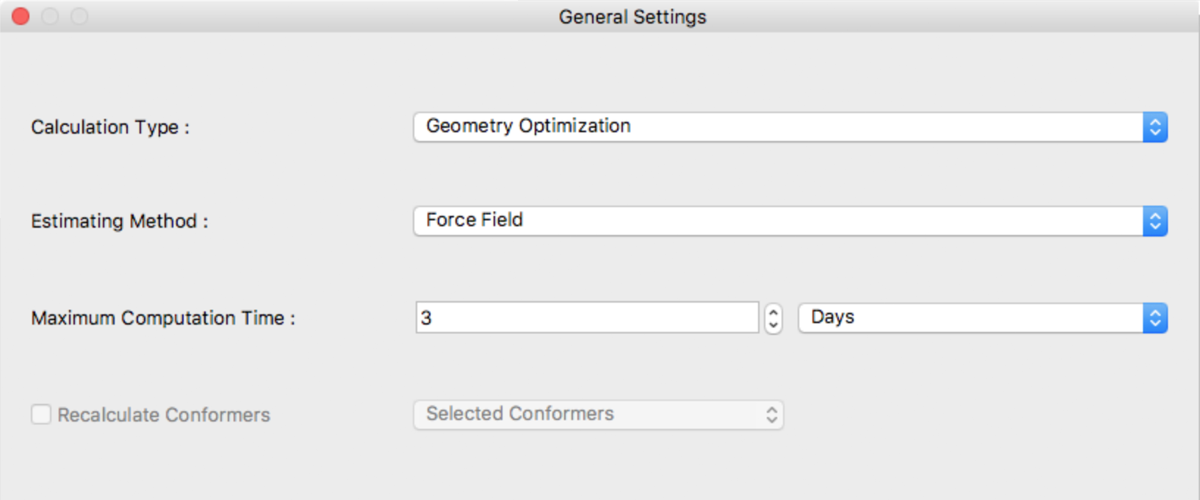
このダイアログでは、実行する計算の内容と制限時間を設定します。
Calculation Type:
「Calculation Type:」からは、以下が選択可能です。
- Geometry Optimization
- 構造最適化のみ
- Conformation Search
- 配座探索+構造最適化
- NMR 3JHH - Coupling Constants
- プロトンNMRの3JHH結合定数を算出
- UV/Vis/CD Spectrum
- UV/Vis/CDスペクトルを算出
- Molecular Crystal
- 結晶の、分子構造・格子定数・分子配向・並進位置の最適化
- Crystal Search
- 結晶構造探索
- Host Ligand Search
- 複数分子間のホストーリガンド探索
- Dynamic Reaction Coordinate
- 動的反応座標を算出
Estimating Method:
「Estimating Method:」では、構造最適化を行う際の手法を指定します。以下の手法が選択可能です:
- Force FIeld
- 分子力場を使用
- External Program
- 外部プログラムの呼び出し(Gaussianに対応)
- Force FIeld and External Program
- 分子力場で最適化後、外部プログラムで引き続き最適化
Maximum Computation Time:
「Maximum Computation Time:」では、シミュレーションの制限時間を設定します。 時間単位は、以下から選択可能です。
- Days
- Hours
- Minutes
Recalculate Conformers
「Recalculate Conformers」では、サーバー上のCONFLEXアプリケーションを利用して、配座の構造の再計算を行います。主に、「UV/Vis/CD」計算に対して利用します。適用範囲は、以下から選択可能です。
- Selected Coformers
- All Conformers
Force Fieldダイアログ
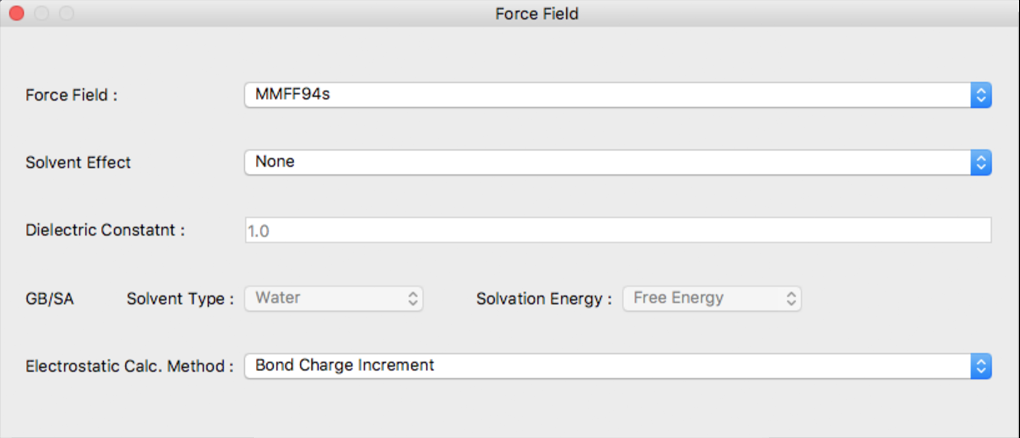
このダイアログでは、構造最適化に利用する力場の設定を行います。
Force Field:
「Force Field:」では、利用する力場を指定します。選択肢は以下のとおりです:
- MMFF94s
- EMM2
Solvent Effect
「Solvent Effect」では、溶媒効果の使用を指示できます。選択肢は以下のとおりです:
- None
- 溶媒効果は使用しません
- Dielectric Constant
- 静電相互作用に比誘電率を適用します
- GB/SA
- Generalized Born / Surface Area法を適用します。MMFF94sのみに対応しています
「Dielectric Constant」を選択した場合、すぐ下の「Dielectric Constant:」ボックスに数値を設定します。
「GB/SA」を選択した場合、溶媒およびエネルギー法をその下のGB/SA設定から選択します。「GB/SA」には、「Solvent Type:」と「Solvent Free Energy:」の設定があります。
「Solvent Type:」では、溶媒の種類を以下から選択します:
- Water(水)
- Octanol(オクタノール)
「Solvation Free Energy:」では、GB/SAによる溶媒和エネルギー解析の手法を選択します。
- Single Point
- Optimization
- Free Energy
詳しくは、CONFLEXマニュアルのキーワードオプションをお読みください。
「Electrostatic Calc. Method:」では、静電相互作用を計算する手法を選択できます。
- Bond Charge Increment
- 結合電荷増分法
- NQEq
- 新規電荷平衡法
- Bond Dipole
Geometry Optimizationダイアログ
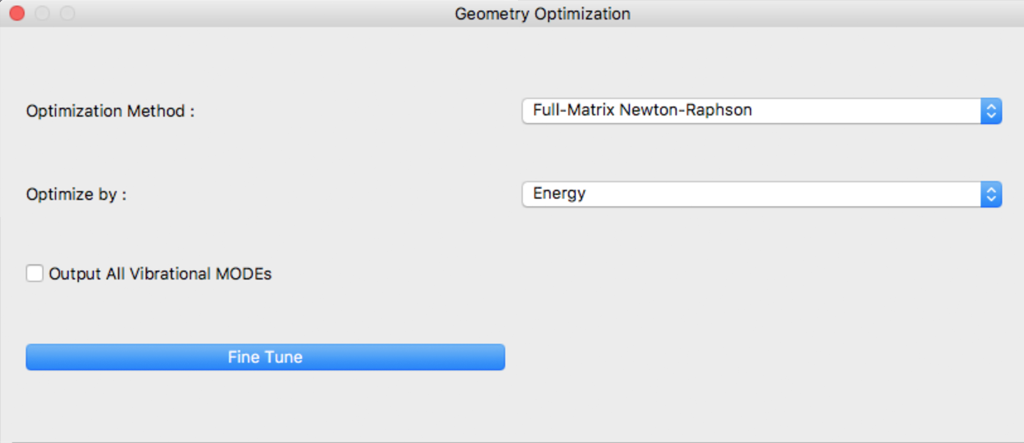
このダイアログでは、構造最適化に関する設定を行います。
Optimization Method:
「Optimization Method:」では、最適化アルゴリズムを選択します。 通常は、「Full-Matrix Newton-Raphson」を使用してください。
- Full-Matrix Newton-Raphson
- No Optimization
- Steepest Descent
- Conjugate Gradient
- Variable Metric
- FAST for large molecule
- PRECISE
- Frontier Mode Following
- Group Optimization
詳しくは、CONFLEXマニュアルのキーワードオプションをお読みください。
Optimize by:
「Optimize by:」では、構造最適化中に最適化する対象を指定します。
- Energy
- Gradient
Output All Vibrational MODEs
「Output All Vibrational MODEs」にチェックを入れると、出力ファイルに全ての振動モードが出力されます。
Fine Tune
「Fine Tune」ボタンをクリックすると、詳細設定ダイアログに表示が切り替わり、より詳細に設定を行えます。
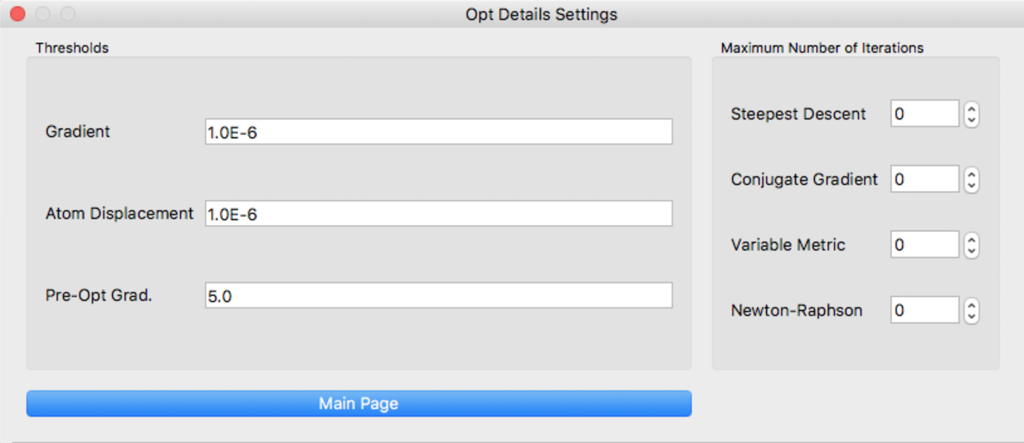
左の「Thresholds」では、計算の終了条件を指定できます。 右の「Maximum Number of Iterations」では、各アルゴリズムの最大繰り返し回数を設定できます。
詳しくは、CONFLEXマニュアルのキーワードオプションをお読みください。
Conformation Searchダイアログ
このダイアログでは、配座探索に関する設定を行います。
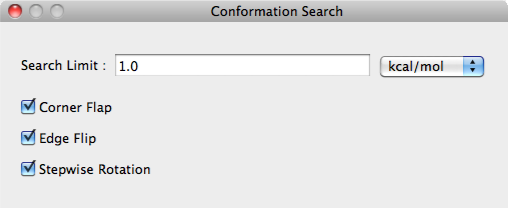
Search Limit:
「Search Limit:」ボックスでは、探索範囲を指定します。単位として、
- kcal/mol
- %
が選択できます。
Corner Flap, Edge Flip, Stepwise Rotation
「Corner Flap」「Edge Flip」「Stepwise Rotation」チェックボックスでは、配座発生の手法を選択します。
詳しくは、CONFLEXマニュアルのキーワードオプションをお読みください。
UV/Vis/CD Spectrumダイアログ
UV/Vis/CD計算の指定は、General Settingsで行います。まず、General Settingsダイアログの「Calculation Type:」から「UV/Vis/CD Spectrum」を選択してください。
これにより、UV/Vis/CDダイアログが表示されます。
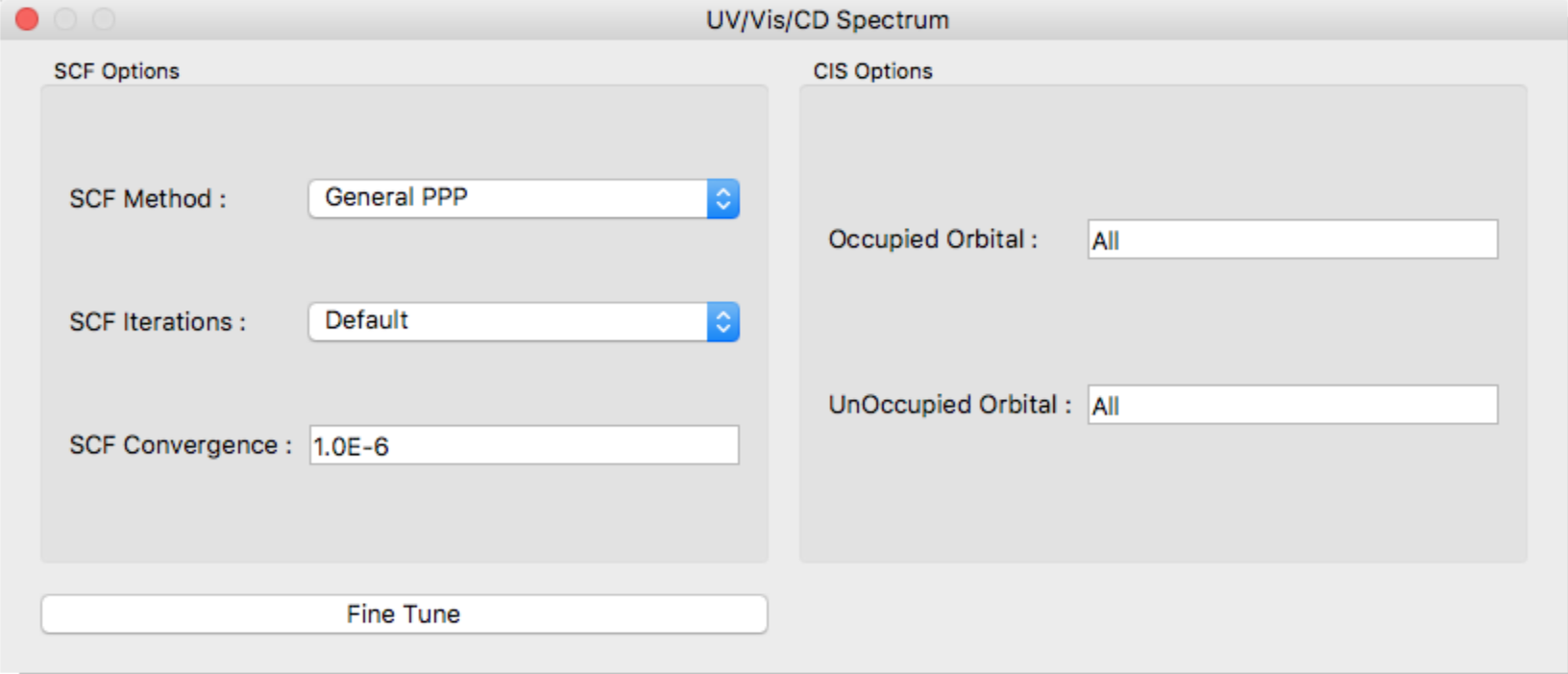
SCF Method:
「SCF Method:」では、SCF計算法を指定します。選択肢は、以下です:
- GeneralPPP
- 一般的なPPP/SCF-MO計算を行う
- Variable Electronegativity
- AllingerによるVariable electronegativity法を用いる
SCF Iterations:
「SCF Iterations:」では、繰り返し最大数を指定します(デフォルト:50)。
SCF Convergence:
「SCF Convergence:」はSCF収束の閾値を設定します。
Occupied Orbital, UnOccupied Orbital
「Occupied Orbital」と「UnOccupied Orbital」は、 一電子励起CI計算に用いる占有軌道および非占有軌道の数を指定します。 デフォルトは、それぞれ「All」となっており、全軌道が対象となります。
ダイアログ下部の「Fine Tune」をクリックすると、詳細設定ダイアログに切り替わります。
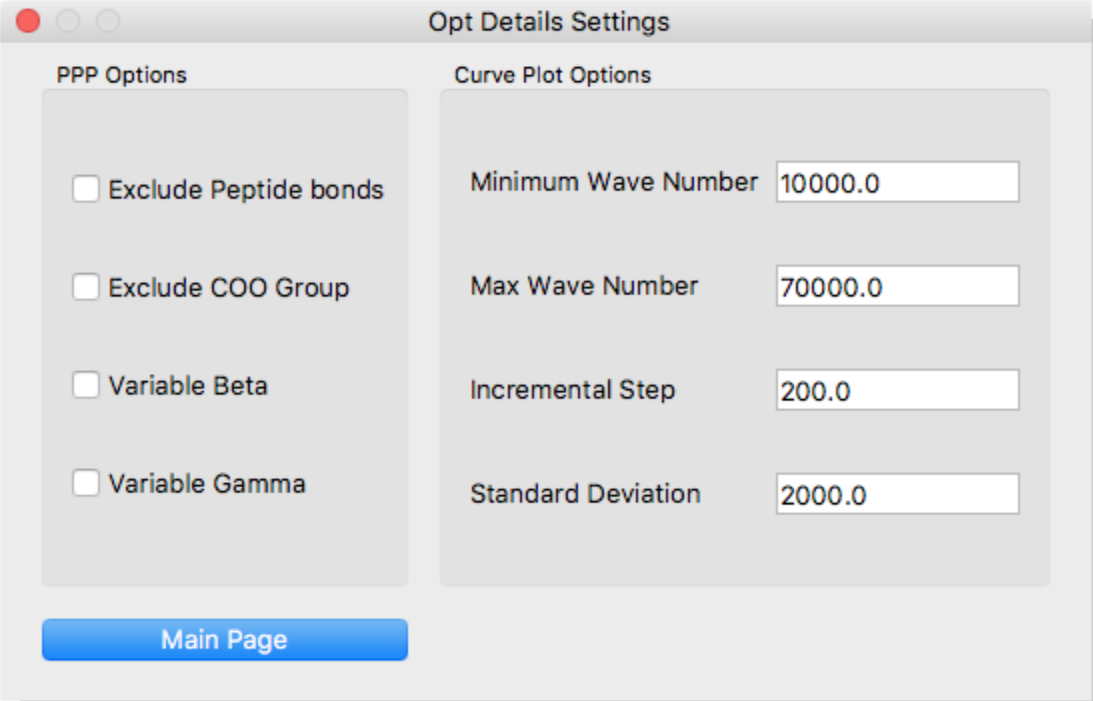
ここでは、以下の設定を行います:
- Exclude Peptide bonds
- ペプチド結合のπ電子を除く
- Exclude COO Group
- カルボキシル基のπ電子を除く
- Variable Beta
- PPP計算のVariable beta拡張法を使用する
- Variable Gamma
- PPP計算のVariable gamma拡張法を使用する
- Minimum Wave Number
- ガウス近似で求めたスペクトルの出力範囲の始点
- Max Wave Number
- ガウス近似で求めたスペクトルの出力範囲の終点
- Incremental Step
- ガウス近似で求めたスペクトルの出力の刻み幅
- Standard Deviation
- ガウス分布の標準偏差を指定します。
ダイアログ下部の「Main Page」ボタンをクリックすると、元のダイアログに戻ります。
NMRダイアログ
NMRの3Jカップリング定数計算の指定を行います。
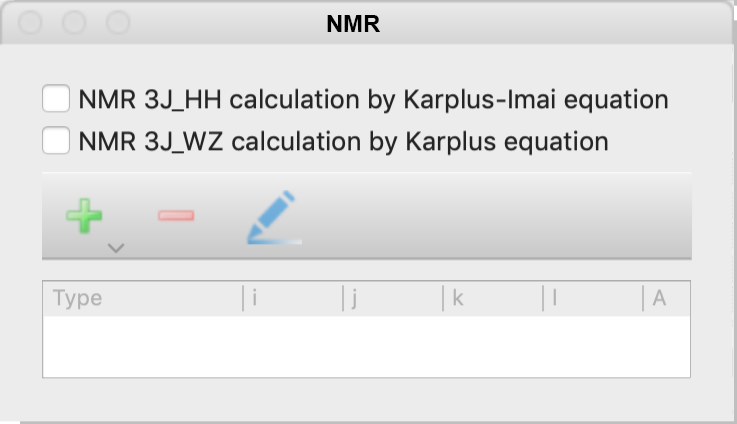
計算内容を、以下から選択できます。
- NMR 3J_HH calculation by Karplus-Imai equation
- Karplus-Imai式によるCsp3-Csp3結合周りの3JHHカップリング定数計算を行います。
- NMR 3J_WZ calculation by Karplus equation
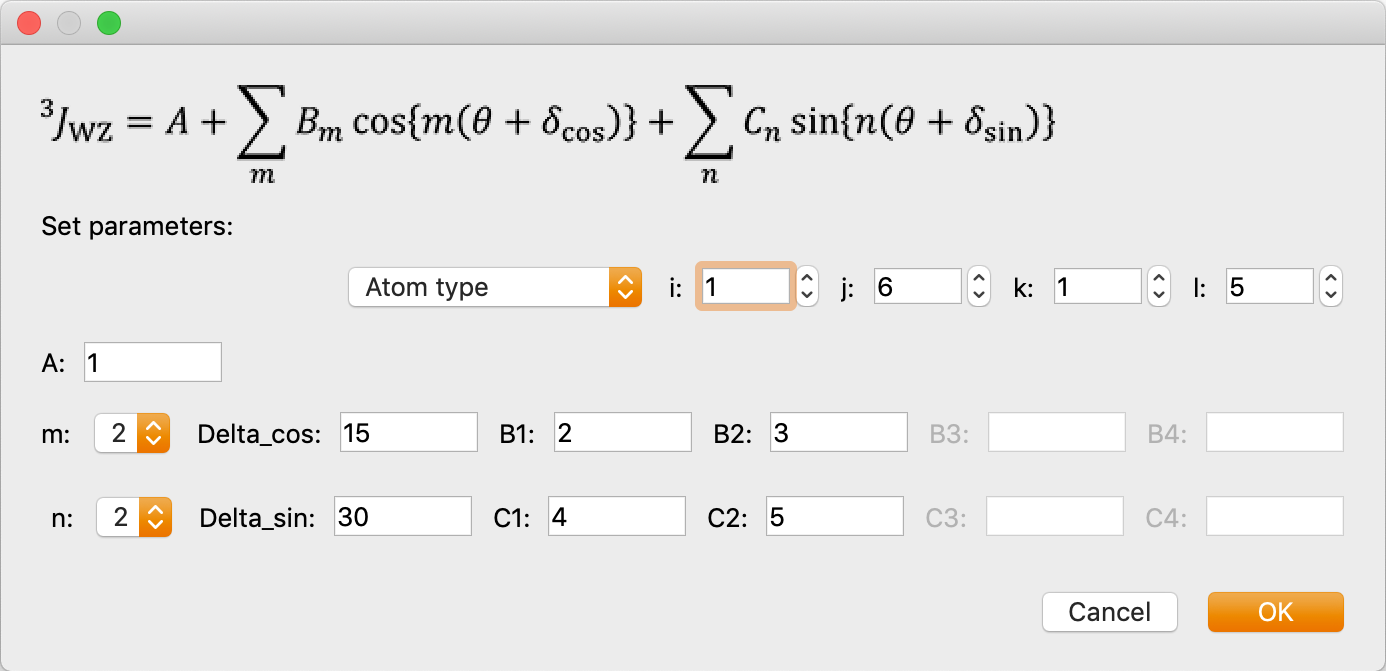
- 原子番号I-J-K-L二面角周りのNMR 3Jカップリング定数を求めます。選択すると、以下のような、パラメーターを入力するダイアログが表示されます。必要なパラメーターを入力してから「OK」をクリックすると、元のダイアログ下部のテーブルに一覧表示されます。また、ダイアログの「+」「-」アイコンや鉛筆のアイコンをクリックすると、パラメーターの追加や削除および入力済みパラメーターの修正ができます。
計算式は、以下です:

この数式の各パラメーターを入力します。
結晶構造最適化
結晶構造最適化の指定は、General Settingsで行います。まず、General Settingsダイアログの「Calculation Type:」から「Molecular Crystal」を選択してください。
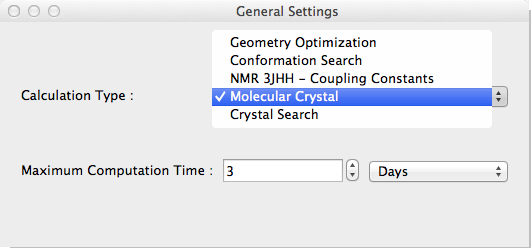
これにより、「Crystal Optimization」ダイアログの設定が可能となります。
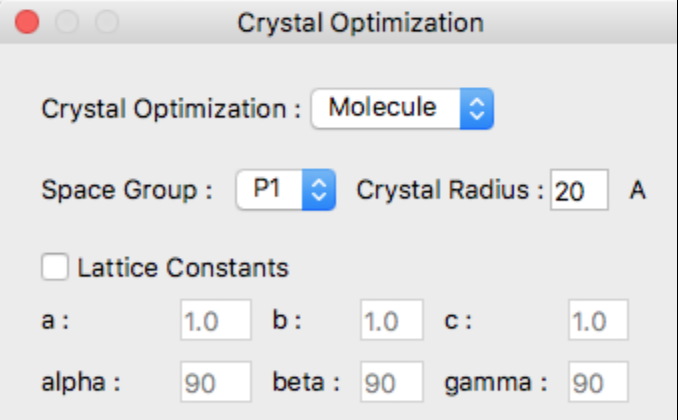
Crystal Optimization:
「Crystal Optimization:」から、最適化手法を選択します。選択肢は、以下です:
- Molecule
- 結晶中の分子構造のみ最適化
- Rigid
- 格子定数・分子配向・並進位置の最適化
- All
- 全て最適化
- None
- 最適化を行わず、プロパティのみ算出
入力ファイルがCIF形式(.cmf, .cif)で、ファイル内に空間群と格子定数の値が含まれる場合、空間群が「Space Group:」に、格子定数の値がそれぞれのボックスに表示されます。その場合、上図の「Lattice Constants」チェックボックスは表示されません。
入力ファイルがMDL-Mol形式(.mol)など結晶構造データを含まない場合、ダイアログ内の空間群は「P1」と表示され格子データa, b, cは1.0、alpha, beta, gammaは90とそれぞれ表示されます。
計算の際、格子定数の初期値は自動で設定されますが、明示的に指定したい場合は、「Lattice Constants」にチェックを入れ、格子定数を入力します。
Space Group:
「Space Group:」は、代表的な以下の空間群が登録されていますので、それらから選択する事が可能です:
- P1
- P-1
- P21/C
- P212121
- P21
- C2/C
- PBCA
- PNA21
- PNMA
- PBCN
これら以外のものを入力したい場合は、「Other...」を選択してください。ダイアログが開きますので、希望の空間群を入力します。
Crystal Radius:
「Crystal Radius」には、有効結晶半径を入力します。この値に従って、球状の結晶が組み上げられ計算が行われます。
計算タイプを「Molecular Crystal」とした場合、もう一つ別のダイアログ「Crystal Property」が表示されます。
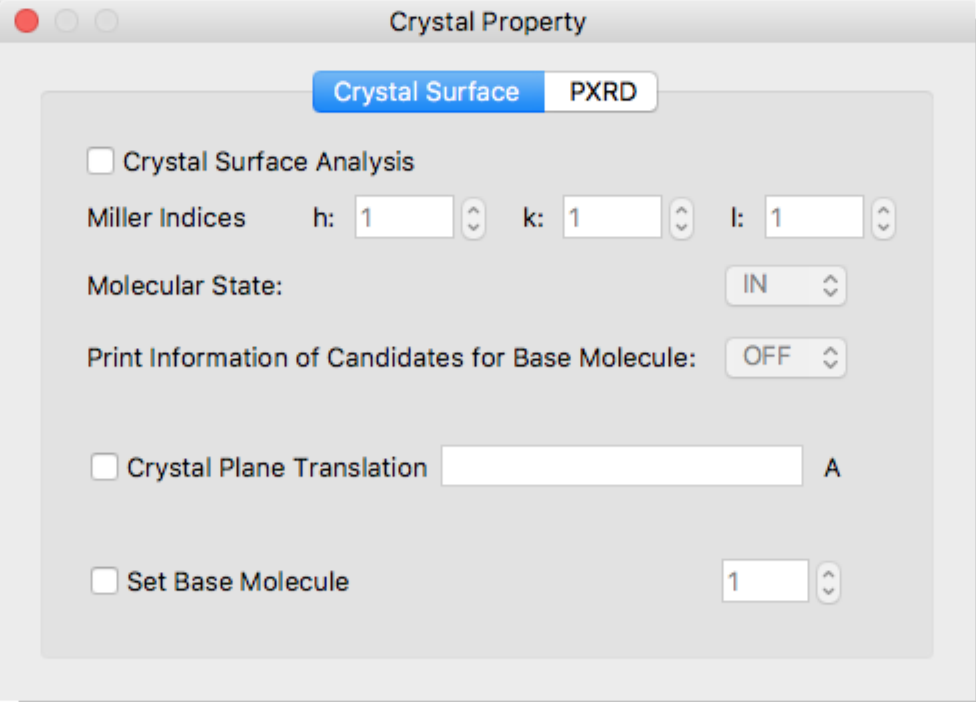
このダイアログでは、結晶表面解析や粉末X線回折パターン計算の詳細設定を行います。
Crystal Surface
「Crystal Surface」タブの「Crystal Surface Analysis」にチェックを入れると、結晶表面解析を実行します。結晶面は、「Miller Indices」の「h:」「k:」「l:」の各ボックスで指定します。
Molecular State:
「Molecular State:」では、エネルギー計算の対象となる分子の状態を以下のどちらかに指定します。
- IN
- 指定結晶面を構成する分子を指定
- ON
- 指定結晶面上に存在する分子を指定
エネルギー計算の対象となる分子の選択は自動で行われます が、明示的に指定したい場合は、「Set Base Molecule」ボックスにチェックを入れ、分子インデックスを指定してください。エネルギー計算の対象として選択できる分子のインデックスは「Print Informa- tion of Candidates for Base Molecule:」を「ON」にすることでbsoファイルに出力されます。
また、界面位置も自動で設定されますが、初期位置から変更したい場合は、「Crystal Plane Translation」ボックスにチェックを入れ並進移動距離を指定します。
PXRD
「Crystal Property」ダイアログ上部の「PXRD」をクリックすると、粉末X線回折パターン計算の設定画面に切り替わります。
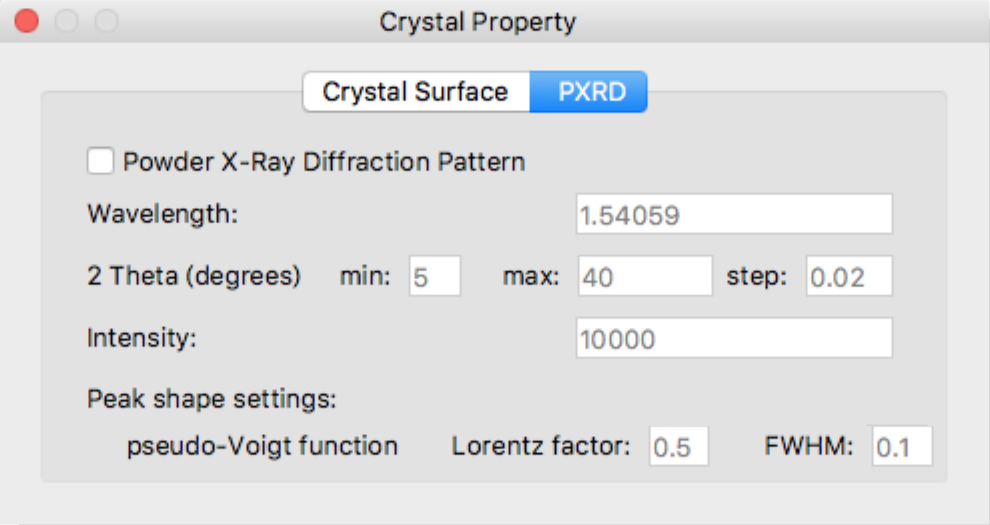
「Powder X-Ray Diffraction Pattern」にチェックを入れると、粉末X線回折パターン計算の詳細設定が行えます。
「PXRD」タブにある各種パラメータの値は必要に応じて変更してください。
- Wavelength
- 放射線の波長を指定
- 2 Theta (degrees)
- 2 Thetaの範囲の最小値・最大値・刻み値を指定
- Intensity
- 回折強度の最大値を指定
- Lorentz factor
- 擬フォークト関数のローレンツ因子を指定
- FWHM
- 擬フォークト関数の半値全幅を指定
結晶構造探索
結晶構造探索を行うには、「General Settings」の「Calculation Type:」 から「Crystal Search」を選択します。
結晶構造の最適化手法を変更したい場合は、「Crystal Optimization」ダイアログの 「Crystal Optimization」で設定を行う事が可能です。
計算設定が「Crystal Search」の場合には、「Crystal Optimization」ダイアログに「Fix Lattice Constants」チェックボックスが表示され、格子定数を固定する指定もできます。結晶構造探索時、格子定数の初期値は自動に設定されますが、特定の格子定数で固定したい場合は、「Fix Lattice Constants」チェックボックスにチェックを入れ、格子定数の値を指定してください。
新たに以下の「Crystal Search」ダイアログが表示されます:
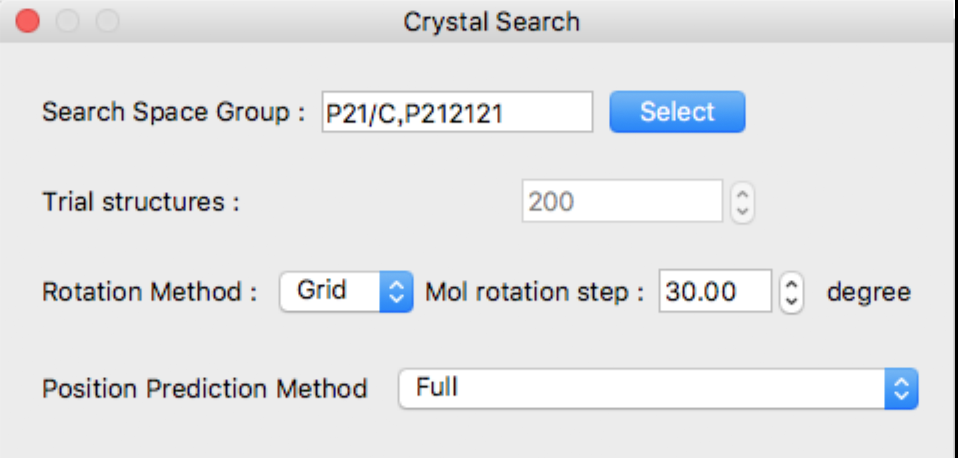 「Crystal Search」ダイアログでは、
「Crystal Search」ダイアログでは、
- 探索に利用する空間群
- 試行構造の数
- 非対称単位を回転させる手法と刻み幅
- 非対称単位の並進位置を求める方法
が選択できます。
空間群は、「Search Space Group:」の横の「Select」ボタンをクリックして以下のダイアログを用いて、選択できます:
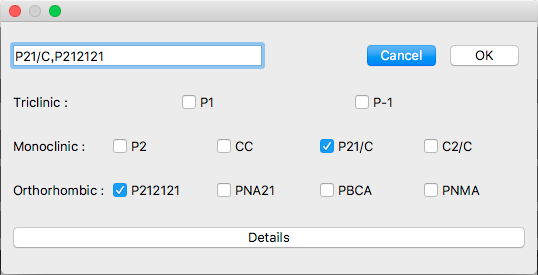
ここには、代表的な10の空間群が表示されていますが、他のものを指定したい場合は、「Detail」ボタンをクリックして、詳細ダイアログを表示させてください。そうすると、全ての空間群を含む、詳細設定ダイアログが表示されます。
元の、簡易ダイアログに戻りたい場合は、ダイアログ下部の「Top 10」ボタンをクリックしてください。
ホスト:リガンド探索
「General Setting」ダイアログの「Calculation Type:」から「Host Ligand Search」を選択すると、分子間の配置探索計算を行えます。
新たに以下の「Host Ligand Search」ダイアログが表示されます:
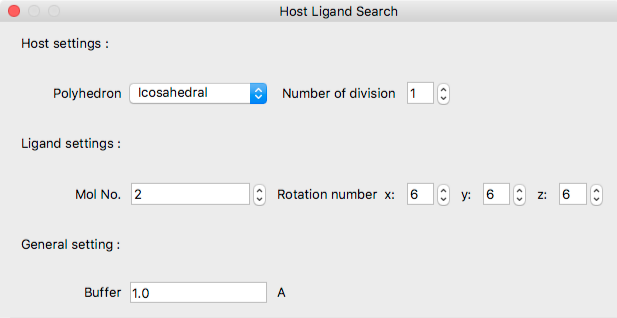
Host Settings:
「Host settings:」の「Polyhedron」からは、ホスト分子の周りの基 準のリガンド分子配置を規定する多面体を選択します。選択肢は、以下です:
- Tetrahedral
- Cube
- Octahedral
- Icosahedral
「Number of division」は、上記多面体の各面の分割数を指定します。ここで指定した数に応じて三角形を各辺の中点で分割し、リガンドの配置位置を増やします。
Ligand Settings:
「Ligand settings:」の「Mol No.」でリンガンドとする分子番号を指定します。
「Rotation number」では、x, y, z各軸周りの回転の刻み数を指定します。「6」の場合、角度は360 / 6 = 60度刻みとなります。
General settings:
「General settings:」の「Buffer」では、初期構造を形成する際の、ホスト分子(群)を囲む球とリガンド分子を囲む球との間の距離を設定します(単位:Å)。
動的反応座標(DRC)法による動力学計算
「General Settings」ダイアログの「Calculation Type:」から「Dynamic Reaction Coordinate」を選択すると、動的反応座標法を利用 した動力学計算を行えます。
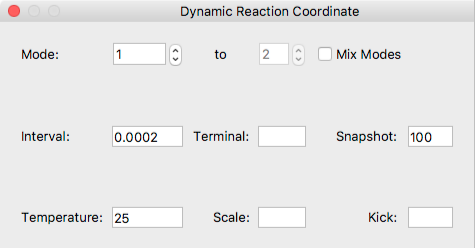
Mode:
「Mode:」では、DRC計算に用いる振動モードを選択できます。
複数の振動モードを混合してDRC計算を行う場合には、「Mix Modes」にチェックを入れ、「to」でモード範囲を指定します。
Interval
「Interval」には、時間の刻み幅を指定します(単位:ps)。
終了時間(単位:ps)を指定したい場合は、「Terminal:」ボックスに書き込みます。
「Snapshot:」には、座標データを出力する頻度を指定します。
Temperature:
「Temperature:」には、DRC計算中の温度(単位:℃)を指定します。
「Scale:」には、振幅のスケーリング・ファクターを指定できます。
「Kick:」に値を指定した場合、振動のスケーリング・ファクターを求める際にこの値が使用されます(単位:kcal/mol)。
外部プログラムを利用した配座探索
「General Setting」ダイアログの「Estimating Method:」で
- External Program
- Force Field and External Program
のいずれかを選択した場合、内蔵の分子力場の代わりに外部の量子化学計算プログラムGaussianを呼び出して、最適化計算や配座探索を実行することができます。
設定は、「External Program」ダイアログで行います。
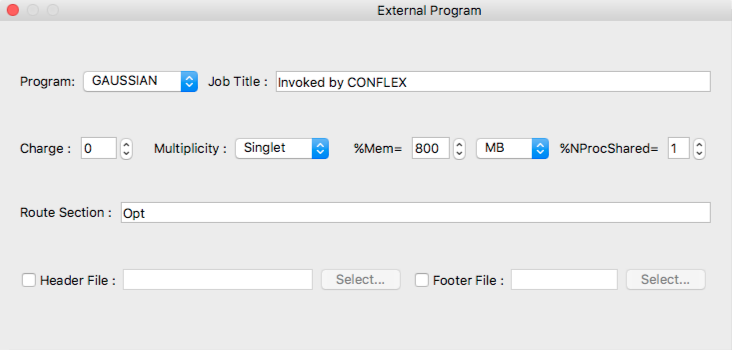
Program:
「Program:」で、外部プログラムを指定します。現バージョンで対応しているのは、GAUSSIANのみです。以下の項目で、Gaussianの計算設定を行います:
- Job Title
- Gaussianの入力ファイルに出力されるジョブのタイトルを指定
- Charge
- 分子系全体の電荷を指定
- Multiplicity
- 分子系のスピン多重度を指定
- %Mem=
- Gaussianの実行に使用するメモリー量の指定。隣のポップアップ・リストから単位を指定します。単位は、
- MB メガ・バイト
- MW メガ・ワード(ワードはバイトの8倍)
- GB ギガ・バイト
- GW ギガ・ワード
- TB テラ・バイト
- TW テラ・ワード
- %NProcShared=
- Gaussian実行時の並列度を指定
- Route Section
- Gaussianの入力ファイルのrouteセクションを指定
Gaussianの計算設定は、同じフォルダー内の別のファイルに記述することも可能です。
別途作成したファイルは、「Header File」ボックスにチェックを入れてから「Select」ボタンをクリックして選択します。
分子構造の下に設定が必要な入力の場合、それを含む別のファイルを作成し、「Footer File:」ボックスにチェックを入れてから「Select」ボタンをクリックして選択します。