結晶表面解析
【計算概要】
結晶表面の解析は、結晶の成長や溶解、昇華など、結晶表面で起こる現象を理解する上で重要です。
CONFLEXは、指定した結晶面の比表面エネルギーや、指定した結晶面における分子の相互作用エネルギーを求めることができます。
【比表面エネルギーの算出】
CONFLEXは、指定結晶面の比表面エネルギーEhklを下記の式より求めます[Kim, Y., Matsumoto, M., Machida, K., Chem. Pharm. Bull. 33(10), 4125 (1985)., 分子力学法 大澤映二(編)町田勝之輔(著)]。
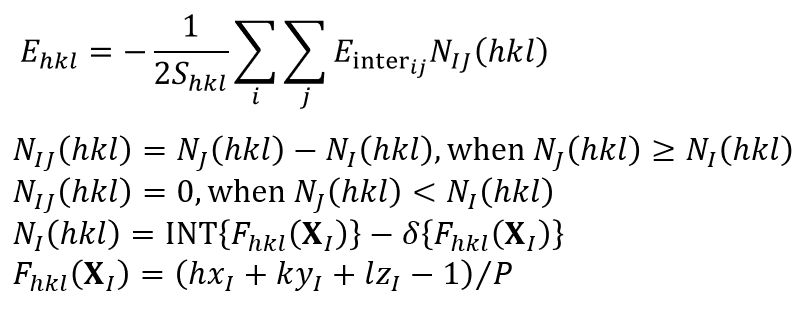
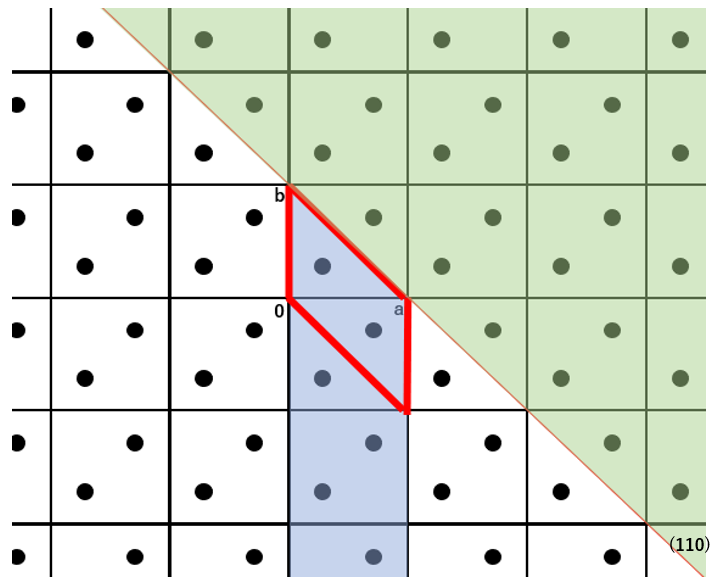
比表面エネルギーの計算式において、Shklは単位格子露出面の面積、Einterijは原子iと原子jとの原子間相互作用エネルギーです。また、iは基本単位格子に含まれるすべての分子の原子をとり、jは原子iを含む分子との最近接分子間距離がカットオフ距離Rcrystal以内の分子の原子をとります。Iは原子iを含む分子の番号をとり、Jは原子jを含む分子の番号をとります。NIJ(hkl)は、結晶を切断する結晶面を隔てて存在する同等なI-J分子対の数です。XIは、分子IのFractional coordinate系で示す中心座標です。Pは、h、k、lの各絶対値の最大公約数です。INT(F)は、実数Fの整数部を意味し、δ(F)は、Fが正のとき0、負のとき1となります。つまり、式における二重和は、結晶面の片側にある1個の単位格子とその背後の柱状部分(青色の領域)に存在するすべての分子と、結晶面の反対側(緑色の領域)に存在するすべての分子との、分子間相互作用エネルギーの総和であり、単位格子当たりの表面エネルギーとなります。このように、単位格子あたりの表面エネルギーを算出し、その露出表面積で割ることで、比表面エネルギーを求めています。なお、比表面エネルギーは単位格子あたりの表面エネルギーを利用するため、空間群P1でのみ計算できます。
【計算の実行方法】
ここでは、マロン酸誘導体の一つであるヒドロキシマロン酸の結晶(Roelofsen, G. et al, Acta Cryst.1978, B34, 2565.)を利用します。
入力ファイル(tartronicacid.cmf)を以下に示します。tartronicacid.cmfファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\plane\in_plane\tartronicacid.cmf)。
tartronicacid.cmfファイル
data_Tartronicacid _symmetry_cell_setting ORTHORHOMBIC _symmetry_space_group_name_H-M 'P212121 ' _ccdc_symmetry_space_group_name P212121 _symmetry_Int_Tables_number 19 loop_ _symmetry_equiv_pos_site_id _symmetry_equiv_pos_as_xyz 1 x,y,z 2 1/2+x,1/2-y,-z 3 -x,1/2+y,1/2-z 4 1/2-x,-y,1/2+z _cell_length_a 4.49400 _cell_length_b 8.81900 _cell_length_c 10.88200 _cell_angle_alpha 90.00000 _cell_angle_beta 90.00000 _cell_angle_gamma 90.00000 _cell_formula_units_Z 4 _cell_volume 431.28180 _exptl_crystal_density_diffrn 1.84821 loop_ _ccdc_atom_site_atom_id_number _atom_site_label _atom_site_type_symbol _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _ccdc_atom_site_symmetry _ccdc_atom_site_base 1 O1 O 1.12990 -0.13910 0.36040 1_555 1 2 O2 O 0.97510 0.09280 0.30700 1_555 2 3 O3 O 1.01480 0.11550 0.66290 1_555 3 4 O4 O 1.13030 -0.12820 0.62810 1_555 4 5 O5 O 0.57240 0.09790 0.48970 1_555 5 6 C1 C 0.97750 -0.01540 0.37600 1_555 6 7 C2 C 0.78810 -0.01520 0.49230 1_555 7 8 C3 C 0.99010 -0.00160 0.60420 1_555 8 9 H1 H 0.66800 -0.11200 0.49600 1_555 9 10 H2 H 0.60500 0.14900 0.45600 1_555 10 11 H3 H 1.23700 -0.13800 0.31000 1_555 11 12 H4 H 1.27100 -0.12000 0.68000 1_555 12 loop_ _atom_id _atom_type _atom_attach_nh _atom_attach_h _atom_charge 1 O 1 1 0 2 O 1 0 0 3 O 1 0 0 4 O 1 1 0 5 O 1 1 0 6 C 3 0 0 7 C 3 1 0 8 C 3 0 0 loop_ _bond_id_1 _bond_id_2 _bond_type_ccdc _bond_environment 1 6 S chain 1 11 S chain 2 6 D chain 3 8 D chain 4 8 S chain 4 12 S chain 5 7 S chain 5 10 S chain 6 7 S chain 7 8 S chain 7 9 S chain
[Interfaceから実行する場合]
tartronicacid.cmfファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
まず、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。
次に、「Crystal Calculation」ダイアログの「Crystal optimization:」のプルダウンメニューから「None」を選択します。結晶構造最適化を行ったうえで、結晶表面解析を行いたい場合は、「ALL」などを選択してください。
次に、「Crystal Property」ダイアログの「Crystal Surface」タブにから、表面解析に関する設定を行います。
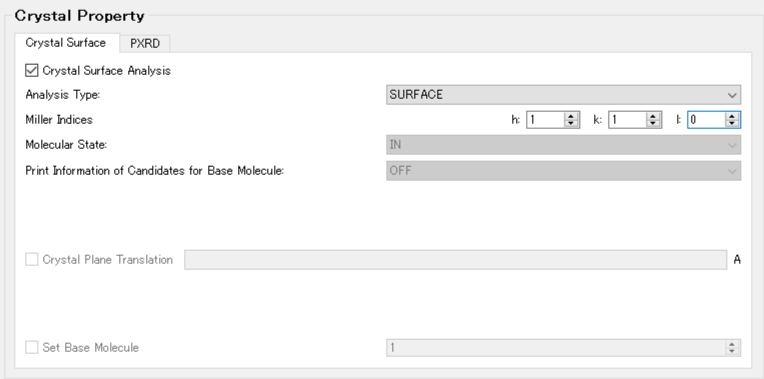
まず、「Crystal Surface Analysis」チェックボックスにチェックを入れます。
次に、「Miller Indices」に解析したい結晶面をhklで指定します。ここでは、h=1, k=1, l=0とします。
設定が終わりましたら、をクリックしてください。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、tartronicacid.iniファイルにキーワードを記述することで行います。
tartronicacid.iniファイル
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,1,0) MAKEP1CELL
「CRYSTAL」は、結晶計算を行うことを示します。
「CRYSTAL_OPTIMIZATION=NONE」は、結晶構造最適化を実施しないことを意味します。結晶構造最適化を行ったうえで、結晶表面解析を行いたい場合は、「NONE」を「ALL」などにしてください。
「CRYSTAL_PLANE=(1,1,0)」は、(110)面の比表面エネルギーを求めることを意味します。
「MAKEP1CELL」は、空間群をP1に変更することを意味します。
tartronicacid.cmfとtartronicacid.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【計算結果】
tartronicacid.bsoに、(110)面に関する比表面エネルギーE110が下記の通り出力されます。
*** SPECIFIC SURFACE ENERGY CALCULATION
* CHANGE TO CUTOFF METHOD IN CALCULATION OF ELECTROSTATIC INTERACTIONS
SURFACE AREA SPECIFIC
(h k l) d_hkl (Ang.) (Ang.**2) SURFACE ENERGY (mJ/m**2)
1 1 0 4.0041 107.7103 216.2389
また、結晶面の片側にある1個の基本単位格子と、その反対側に存在するカットオフ距離Rcrystal以内の分子で構成した半球を含むモデルのpdbファイルが得られます。ファイル名のフォーマットは、「(入力ファイル名)_hkl.pdb」です。tartronicacid_110.pdbファイルをCONFLEX Interfaceで開くことでモデルを可視化できます。
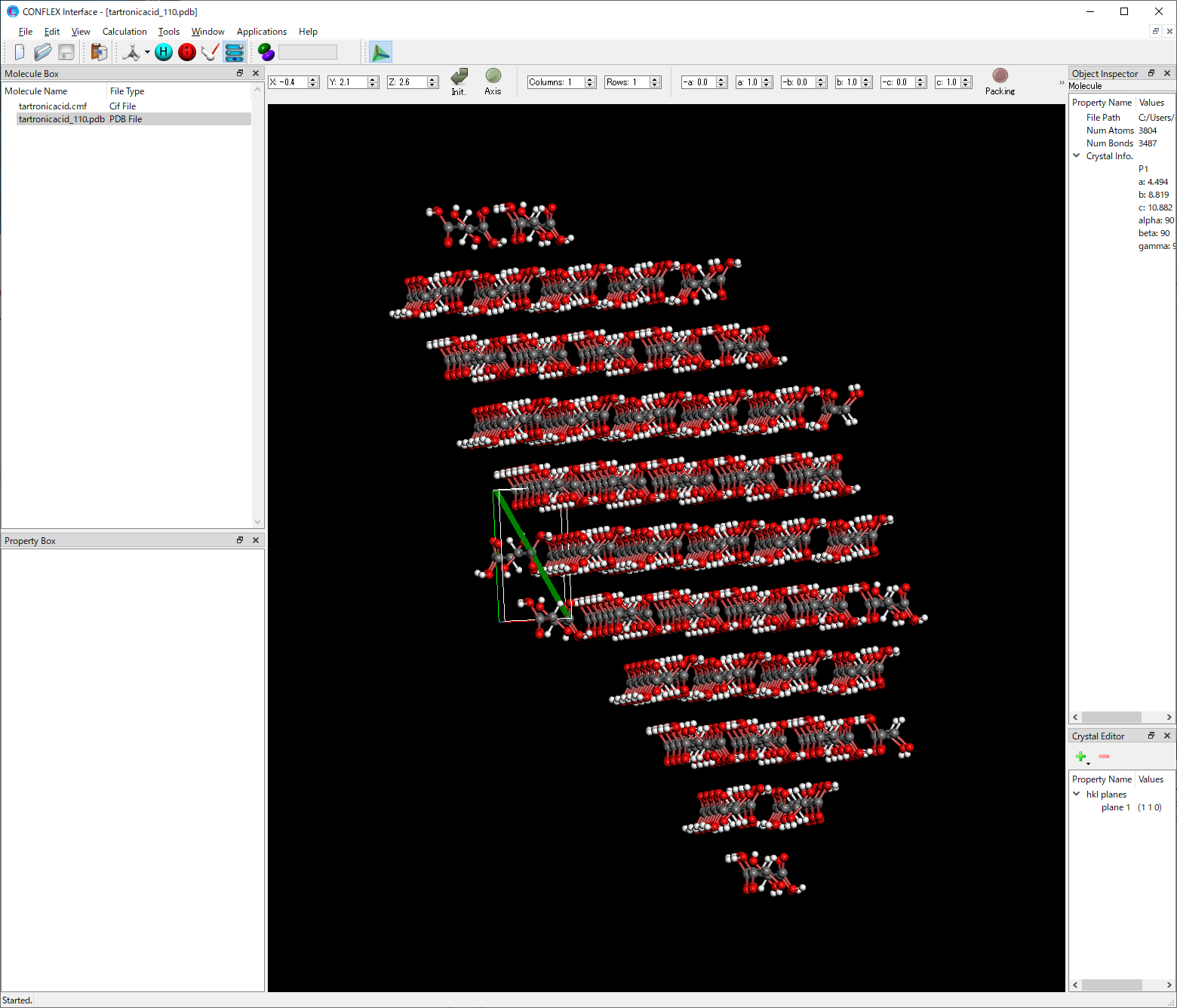
【分子の相互作用エネルギーの算出】
(100)面を指定した場合、CONFLEXは下図に示すように、(100)面を露出させた半球結晶を構築し、(100)面を構成する分子、あるいはその結晶面上に存在する分子の分子間相互作用エネルギーの和を求めることができます。なお、半球の半径は設定したカットオフ距離になります。
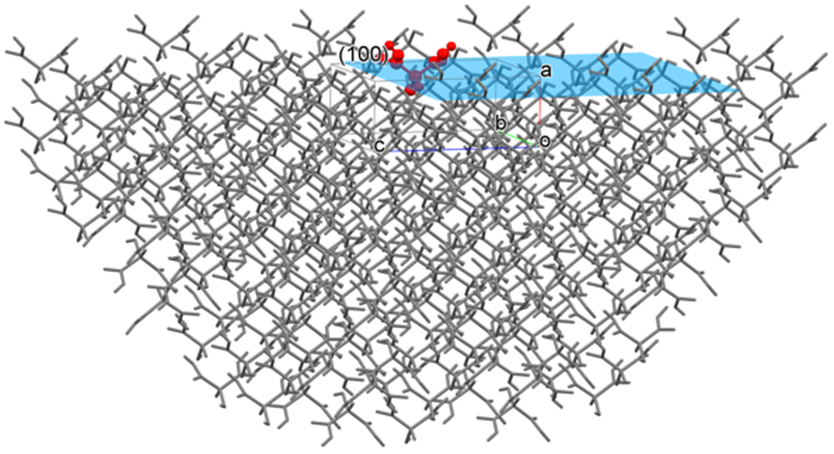

ここで、Emolは露出させた結晶面の中心に存在する分子(上図、赤色の分子。以下、基本分子と呼ぶ)の分子間相互作用エネルギーの和であり、Nは基本分子の総原子数、Mは基本分子以外の半球結晶に含まれる全分子の総原子数です。
【計算の実行方法】
ここでは、マロン酸誘導体の一つであるヒドロキシマロン酸の結晶(Roelofsen, G. et al, Acta Cryst.1978, B34, 2565.)を利用します。
入力ファイル(tartronicacid.cmf)を以下に示します。tartronicacid.cmfファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\plane\in_plane\tartronicacid.cmf)。
tartronicacid.cmfファイル
data_Tartronicacid _symmetry_cell_setting ORTHORHOMBIC _symmetry_space_group_name_H-M 'P212121 ' _ccdc_symmetry_space_group_name P212121 _symmetry_Int_Tables_number 19 loop_ _symmetry_equiv_pos_site_id _symmetry_equiv_pos_as_xyz 1 x,y,z 2 1/2+x,1/2-y,-z 3 -x,1/2+y,1/2-z 4 1/2-x,-y,1/2+z _cell_length_a 4.49400 _cell_length_b 8.81900 _cell_length_c 10.88200 _cell_angle_alpha 90.00000 _cell_angle_beta 90.00000 _cell_angle_gamma 90.00000 _cell_formula_units_Z 4 _cell_volume 431.28180 _exptl_crystal_density_diffrn 1.84821 loop_ _ccdc_atom_site_atom_id_number _atom_site_label _atom_site_type_symbol _atom_site_fract_x _atom_site_fract_y _atom_site_fract_z _ccdc_atom_site_symmetry _ccdc_atom_site_base 1 O1 O 1.12990 -0.13910 0.36040 1_555 1 2 O2 O 0.97510 0.09280 0.30700 1_555 2 3 O3 O 1.01480 0.11550 0.66290 1_555 3 4 O4 O 1.13030 -0.12820 0.62810 1_555 4 5 O5 O 0.57240 0.09790 0.48970 1_555 5 6 C1 C 0.97750 -0.01540 0.37600 1_555 6 7 C2 C 0.78810 -0.01520 0.49230 1_555 7 8 C3 C 0.99010 -0.00160 0.60420 1_555 8 9 H1 H 0.66800 -0.11200 0.49600 1_555 9 10 H2 H 0.60500 0.14900 0.45600 1_555 10 11 H3 H 1.23700 -0.13800 0.31000 1_555 11 12 H4 H 1.27100 -0.12000 0.68000 1_555 12 loop_ _atom_id _atom_type _atom_attach_nh _atom_attach_h _atom_charge 1 O 1 1 0 2 O 1 0 0 3 O 1 0 0 4 O 1 1 0 5 O 1 1 0 6 C 3 0 0 7 C 3 1 0 8 C 3 0 0 loop_ _bond_id_1 _bond_id_2 _bond_type_ccdc _bond_environment 1 6 S chain 1 11 S chain 2 6 D chain 3 8 D chain 4 8 S chain 4 12 S chain 5 7 S chain 5 10 S chain 6 7 S chain 7 8 S chain 7 9 S chain
[Interfaceから実行する場合]
tartronicacid.cmfファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
まず、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから「Molecular Crystal」を選択します。
次に、「Crystal Calculation」ダイアログの「Crystal optimization:」のプルダウンメニューから「None」を選択します。結晶構造最適化を行ったうえで、結晶表面解析を行いたい場合は、「ALL」などを選択してください。
次に、「Crystal Property」ダイアログの「Crystal Surface」タブにから、表面解析に関する設定を行います。
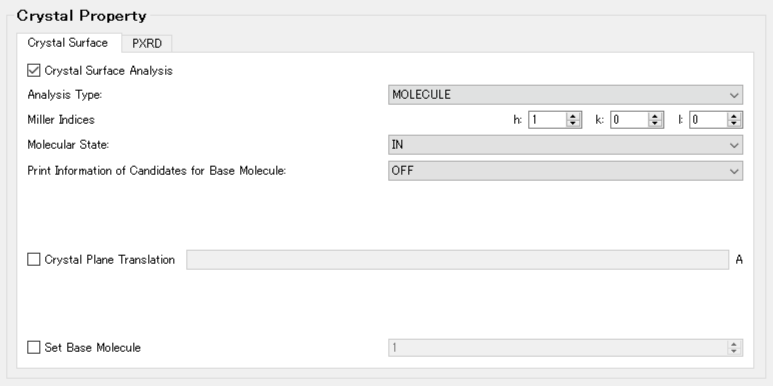
まず、「Crystal Surface Analysis」チェックボックスにチェックを入れます。また、「Analysis Type:」を「MOLECULE」に変更します。
次に、「Miller Indices」に解析したい結晶面をhklで指定します。ここでは、h=1, k=0, l=0とします。
「Molecular State:」の「IN」は、(100)面を構成する分子のEmolを求めることを意味します。
設定が終わりましたら、をクリックしてください。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、tartronicacid.iniファイルにキーワードを記述することで行います。
tartronicacid.iniファイル
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,0,0) CRYSTAL_PLANE_ANALYSIS_TYPE=MOLECULE
「CRYSTAL」は、結晶計算を行うことを示します。
「CRYSTAL_OPTIMIZATION=NONE」は、結晶構造最適化を実施しないことを意味します。結晶構造最適化を行ったうえで、結晶表面解析を行いたい場合は、「NONE」を「ALL」などにしてください。
「CRYSTAL_PLANE=(1,0,0)」は、(100)面の解析を行うことを意味します。ここでは、(100)面を構成する分子のEmolを求めます。
「CRYSTAL_PLANE_ANALYSIS_TYPE=MOLECULE」は、分子の相互作用エネルギーを計算することを意味します。
tartronicacid.cmfとtartronicacid.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par tartronicacidenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【計算結果】
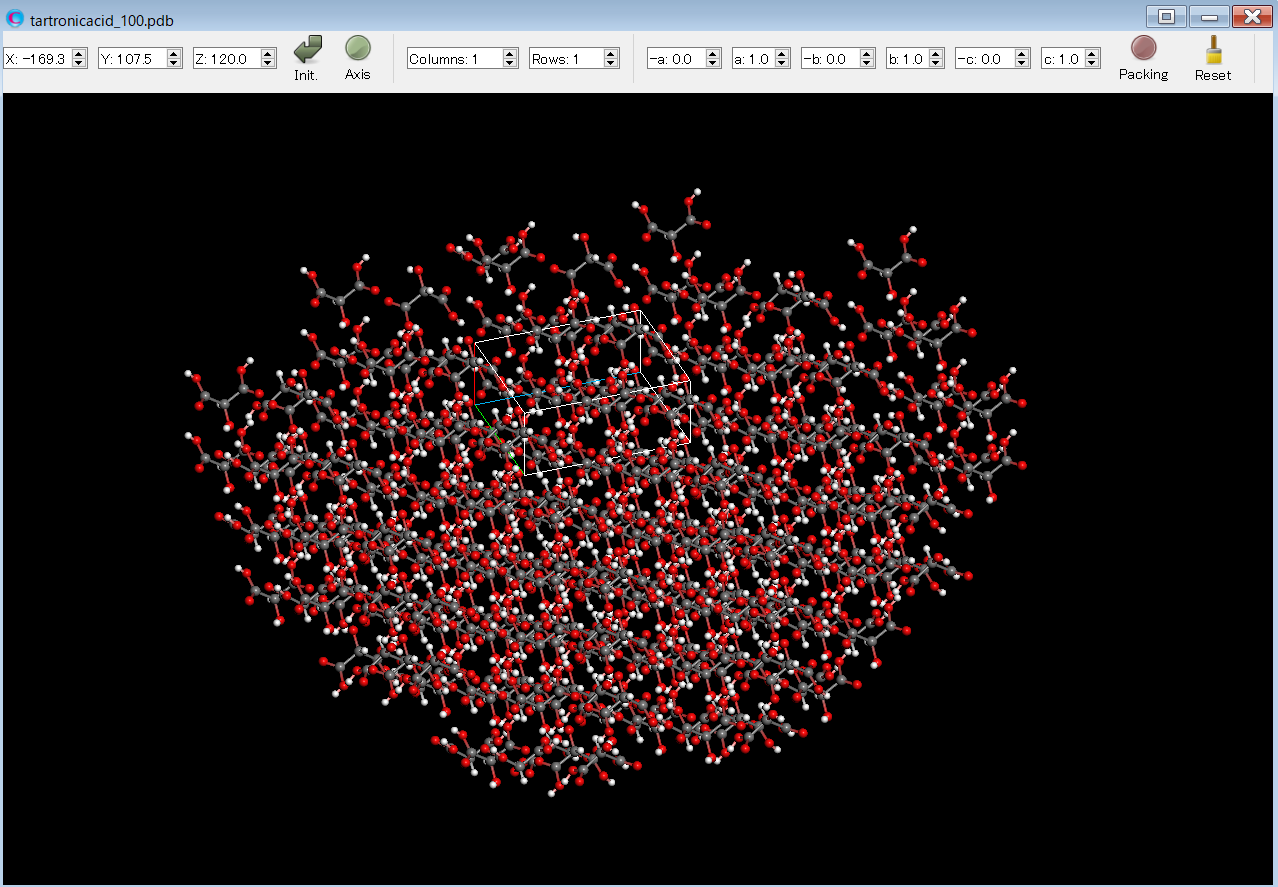
計算終了後、tartronicacid_100.pdbファイルが出力されます。これは(100)面を露出させた半球結晶のpdbファイルです。ファイル名のフォーマットは、「(入力ファイル名)_hkl.pdb」です。tartronicacid_100.pdbファイルをCONFLEX Interfaceで開くことで、半球結晶を可視化できます。
tartronicacid.bsoには下記の通り、基本分子に関するEmolが出力されます。
* SUM OF INTERMOLECULAR INTERACTION ENERGIES OF MOLECULE
[IN] THE CRYSTAL PLANE
* CHANGE TO CUTOFF METHOD IN CALCULATION OF ELECTROSTATIC INTERACTIONS
ENERGY
H K L (KCAL/MOL)
1 0 0 -10.7323
【エネルギーの横にあるアルファベット表記について】
tartronicacid.cmfを入力ファイルとして、ヒドロキシマロン酸結晶の(010)面を解析します。この際、結晶構造最適化は行っていません。
tartronicacid.bsoには下記の通り、基本分子に関するEmolが出力されます。
* SUM OF INTERMOLECULAR INTERACTION ENERGIES OF MOLECULE
[IN] THE CRYSTAL PLANE
* CHANGE TO CUTOFF METHOD IN CALCULATION OF ELECTROSTATIC INTERACTIONS
ENERGY
H K L (KCAL/MOL)
0 1 0 -39.2094 a
0 1 0 -38.4304 b
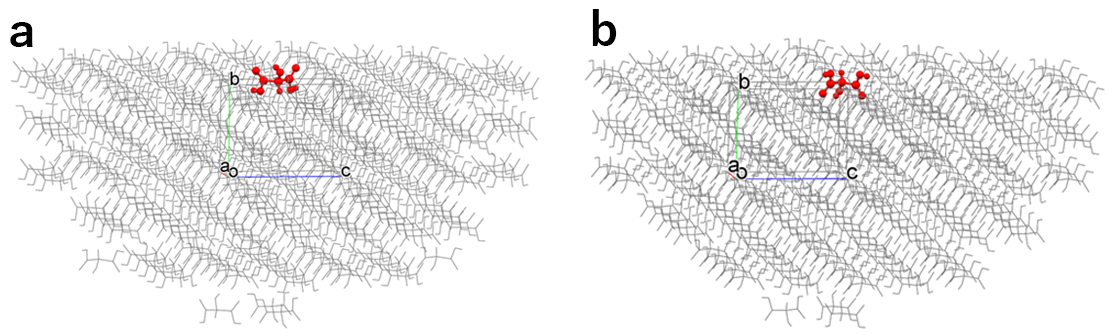
基本分子はユーザーが指定しない限りプログラムが自動に決定しますが、指定結晶面において複数のユニークな分子が存在する場合、それぞれの分子を基本分子としたときのエネルギー値とpdbファイルがアルファベットにより区別され出力されます。aとbは両者ともに(010)面を露出させた半球結晶ですが、エネルギー計算における基本分子が異なります。
【「Molecular State:」の「IN」と「ON」の違い】
「Crystal Property」ダイアログの「Crystal Surface」タブにおいて、「Molecular State:」を「IN」とした場合と「ON」にした場合で、下図のように基本分子の状態が異なります。
コマンドラインからの実行では、iniファイル内に「CRYSTAL_PLANE_STATE=IN」と記述するか、「CRYSTAL_PLANE_STATE=ON」と記述するかに対応します。
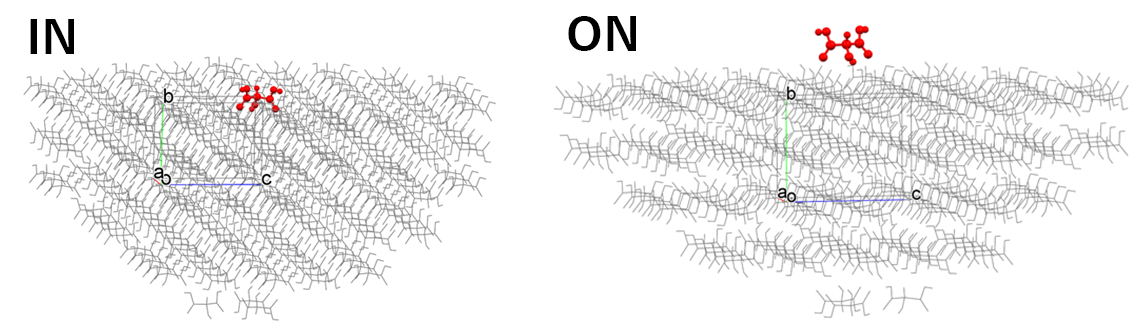
「IN」は指定結晶面を構成する分子が基本分子として選択され、
「ON」は指定結晶面上に存在する分子が基本分子として選択され、Emolが計算されます。
【「Crystal Plane Translation」の使い方】
tartronicacid.cmfを入力ファイルとして、ヒドロキシマロン酸結晶の(001)面を解析します。この際、結晶構造最適化は行っていません。CONFLEXは、下図のような半球結晶を構築しEmolを求めます。
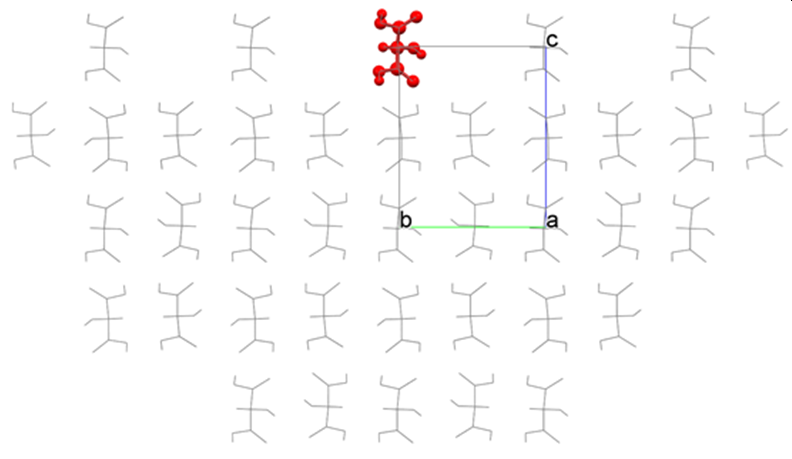
半球結晶の界面の位置はプログラムが自動に決定しますが、「Crystal Property」ダイアログの「Crystal Surface」タブにある「Crystal Plane Translation」の値を設定することで変更できます。なお上図において、結晶面の位置が“0”であり、上方が“+”、下方が”-”方向になります。
例えば、「Crystal Plane Translation」の値を「1.0」に設定し、界面を1.0 Ang.上方に移動させます。
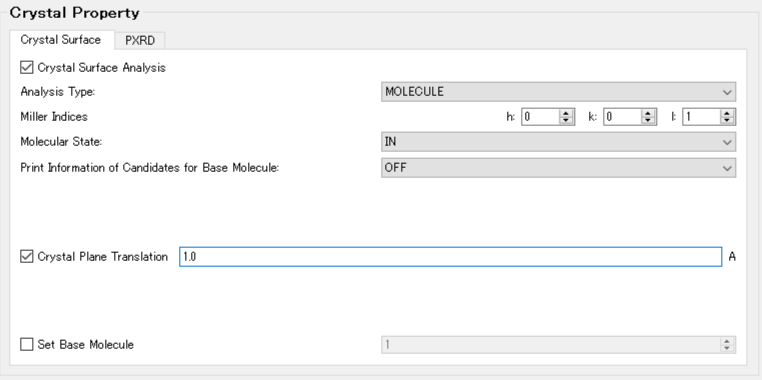
コマンドラインからの実行では、iniファイル内に「CRYSTAL_PLANE_TRANS=1.0」を記述することに対応します。
tartronicacid.iniファイル
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(0,0,1) CRYSTAL_PLANE_TRANS=1.0
その結果、下図のような半球結晶を構築しEmolを求めるようになります。
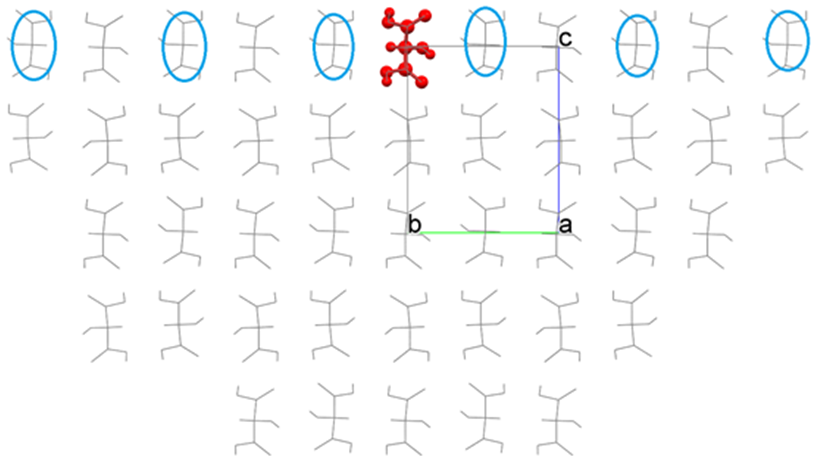
界面を移動させる前と後を比較すると、界面の位置を変更したことで、青丸で示した分子が相互作用エネルギー計算に含まれることがわかります。必要に応じて、界面の位置を変更してください。
【「Set Base Molecule」の使い方】
基本分子はプログラムが自動に決定しますが、「Crystal Property」ダイアログの「Crystal Surface」タブにある「Set Base Molecule」を設定することで変更できます。
ここでは、tartronicacid.cmfを入力ファイルとして、ヒドロキシマロン酸結晶の(100)面の解析を例に説明します。
まず、基本分子の候補となる分子に関する情報を得るため、「Crystal Property」ダイアログの「Crystal Surface」タブの「Print Information of Candidates for Base Molecule:」をONにして、一度、計算を実行します。
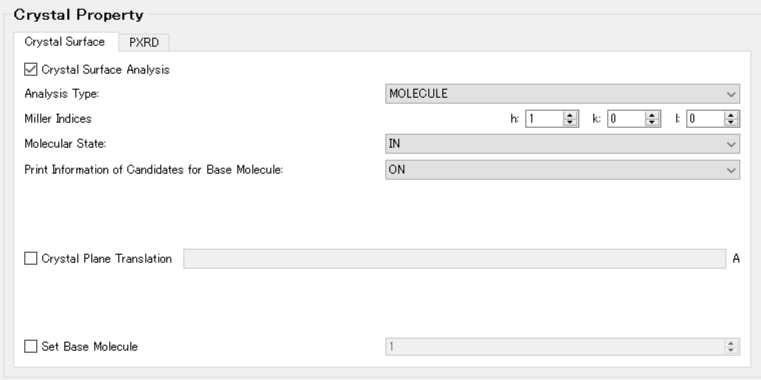
コマンドラインからの実行の場合、iniファイルに「CRYSTAL_PLANE_PRINT=ON」を追加して、一度、計算を実行します。
tartronicacid.iniファイル
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,0,0) CRYSTAL_PLANE_PRINT=ON
tartronicacid.bsoには、下記のように、候補となる分子のインデックス(IDX)と、結晶面の位置を“0”、上方が“+”、下方を“-”方向とした場合の分子の重心位置(POSITION)が出力されます。
IDX POSITION (ANG.) 1 0.224367 2 0.224367 3 -0.224367 4 -0.224367 5 2.022633 6 -2.022633 7 -2.022633 8 -2.022633 9 -2.022633 10 2.471367
例えば、IDX=6の分子を選択する場合、「Crystal Property」ダイアログの「Crystal Surface」タブの「Set Base Mecule」を「6」に設定します。
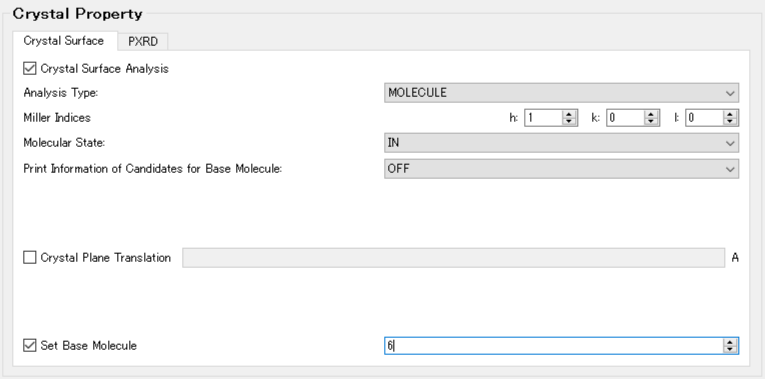
コマンドラインからの実行の場合、iniファイルに「CRYSTAL_PLANE_BASE=6」を追加して計算を実行します。
tartronicacid.iniファイル
CRYSTAL CRYSTAL_OPTIMIZATION=NONE CRYSTAL_PLANE=(1,0,0) CRYSTAL_PLANE_BASE=6
デフォルトでは、左図の半球結晶モデルが構築されEmolが計算されますが、「Set Base Molecule」を「6」とし、基本分子を変更したことで、右図の半球結晶モデルが構築されEmolが計算されるようになります。
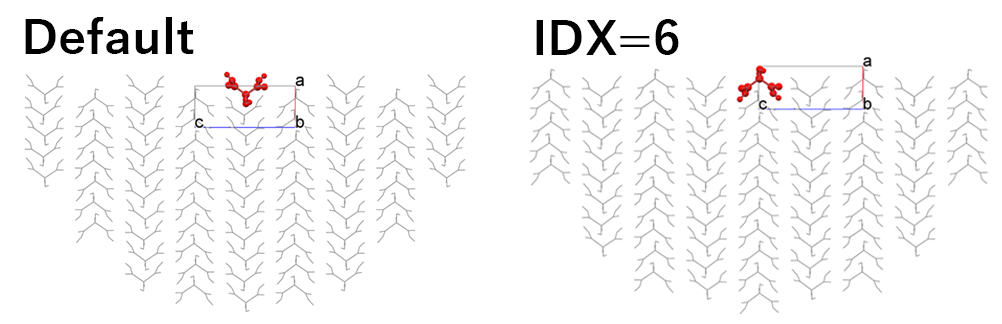
左図の半球結晶モデルの場合、Emolは、-10.73 kcal/molであり、
右図の半球結晶モデルの場合、Emolは、-34.15 kcal/molです。