ホスト-リガンド配位探索
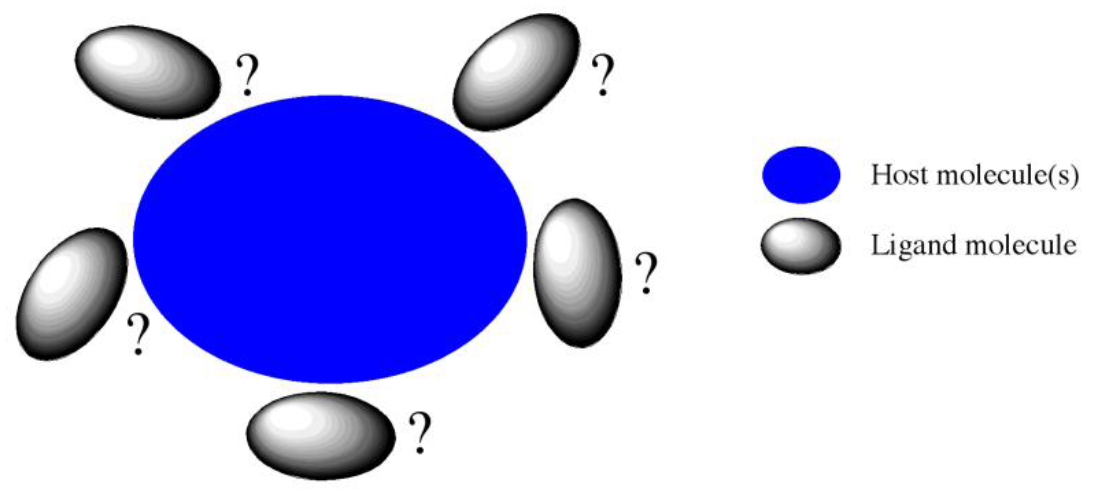
ある分子または分子集合体に対して、他のイオン種や分子がどの位置にどのような向きで配位するのかを探索し(図1)それぞれの安定性を評価することは、錯体や分子クラスターの安定構造を特定するのに有用です。
CONFLEXに搭載された「ホスト−リガンド配位探索(Host-Ligand Coordination Search)」機能により、分子または分子集合体を”ホスト”と指定し、その周囲にイオン種や分子を自動的に配置しそれぞれ構造最適化やエネルギー計算を行うことが可能です。この機能を利用して安定なクラスター構造を見出すことで、分子認識やホスト−ゲスト化学などの超分子化学分野への応用も期待できます。
【計算手法の概要】
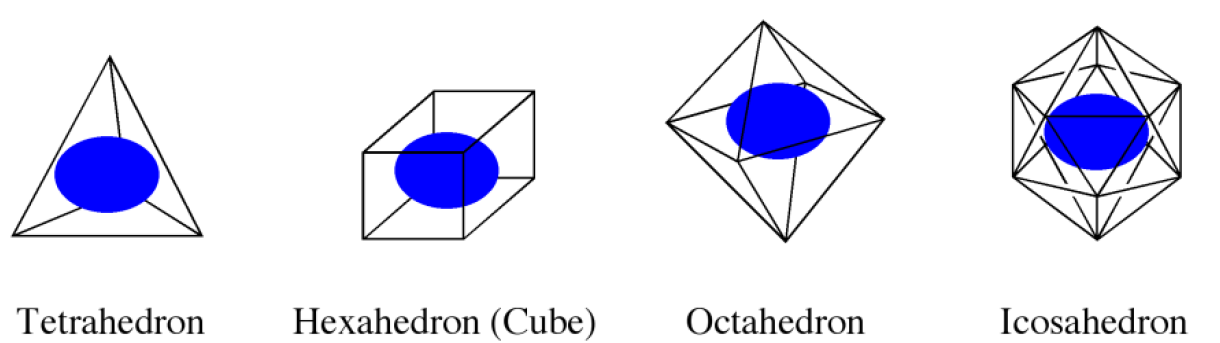
まずホストとなる分子(群)を囲む正多面体を設定します。CONFLEXで用意している正多面体は図2に示す4種類で、デフォルトでは正二十面体(Icosahedron)が選択されます。
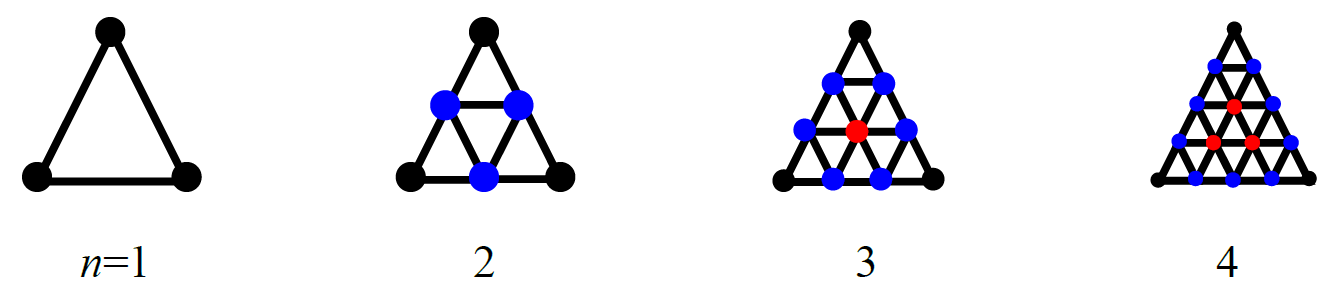
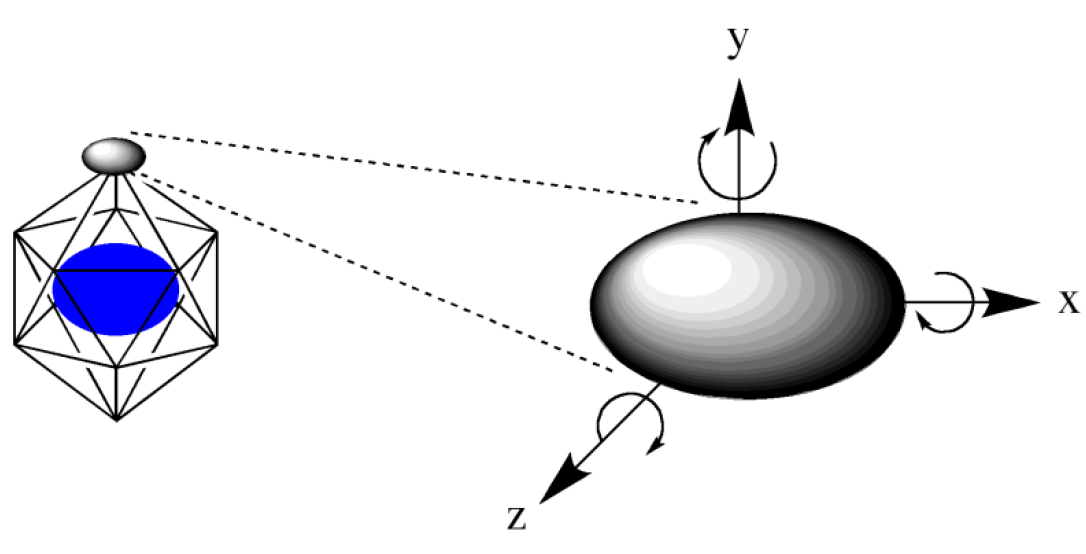
これら多面体の各頂点に、リガンドとなる分子を配置します。配置場所をより細かくしたい場合は、「HLSEARCH_HOST_NDIV=n」により各面を分割することで配置点の数を増やします(下図)。
最後に、配置したリガンド分子をx,y,z軸周りにそれぞれ回転させて初期配置構造を作り(下図)、構造最適化後得られた構造をエネルギー順に出力します。リガンドを回転させる角度は「HLSEARCH_LIGAND_ROT=(l,m,n)」で指定でき、デフォルトでは360/6=60°刻みです(l=m=n=6)。これを例えば45°刻みにする場合は「HLSEARCH_LIGAND_ROT=(8,8,8)」とします。
【酢酸二量体のエネルギー極小構造の探索】
酢酸の二量体(下図)について、それぞれがどのような位置関係・配向にある時にエネルギー極小に成り得るのかを、ホスト−リガンド配位探索により求めます。
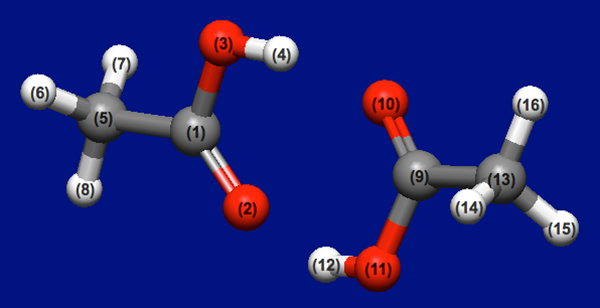
酢酸2分子の入力構造データ(acetic_acid_dimer.mol)
acetic_acid_dimer.mol
16 14 0 0 0 0 0 0 0 0 0 0
0.9628 -0.5511 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1931 -0.5511 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.3335 0.6511 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.9803 1.3755 -0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.0286 -1.7296 0.0001 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6441 -1.6778 0.8738 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.6442 -1.6778 -0.8735 H 0 0 0 0 0 0 0 0 0 0 0 0
0.5138 -2.6519 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
3.5500 2.4393 0.3355 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4107 2.2978 -0.1068 O 0 0 0 0 0 0 0 0 0 0 0 0
4.2100 1.3256 0.7422 O 0 0 0 0 0 0 0 0 0 0 0 0
3.6576 0.5365 0.6183 H 0 0 0 0 0 0 0 0 0 0 0 0
4.3924 3.7159 0.5151 C 0 0 0 0 0 0 0 0 0 0 0 0
4.6407 3.8369 1.5488 H 0 0 0 0 0 0 0 0 0 0 0 0
5.2907 3.6344 -0.0604 H 0 0 0 0 0 0 0 0 0 0 0 0
3.8309 4.5636 0.1817 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0 0 0 0
1 3 1 0 0 0 0
1 5 1 0 0 0 0
3 4 1 0 0 0 0
5 6 1 0 0 0 0
5 7 1 0 0 0 0
5 8 1 0 0 0 0
9 10 2 0 0 0 0
9 11 1 0 0 0 0
9 13 1 0 0 0 0
11 12 1 0 0 0 0
13 14 1 0 0 0 0
13 15 1 0 0 0 0
13 16 1 0 0 0 0
M END
[Interfaceから実行する場合]
acetic_acid_dimer.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
詳細設定ダイアログが表示されます。
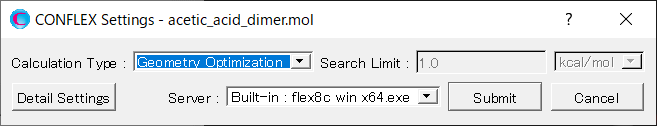
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」 のプルダウンメニューから「Host Ligand Search」を選択します。
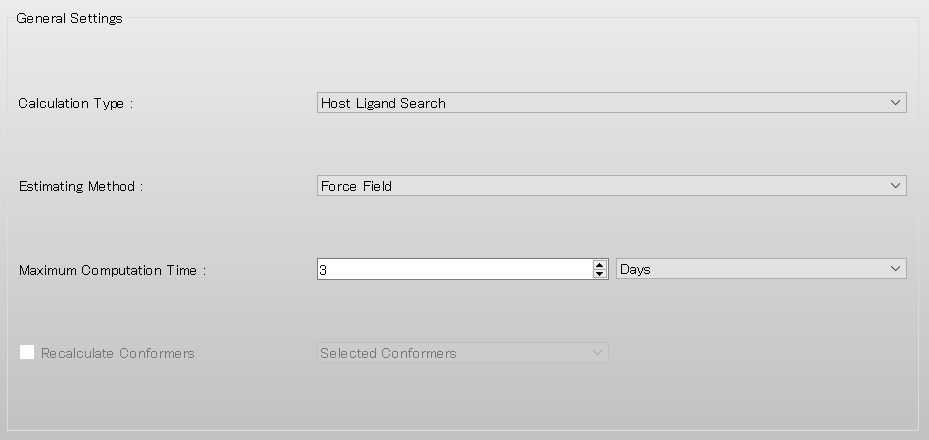
設定が終わりましたら、をクリックします。ホスト−リガンド配位探索が行われます。
[コマンドラインから実行する場合]
計算設定は、acetic_acid_dimer.iniファイルにキーワードを記述することで行います。
acetic_acid_dimer.iniファイル
MMFF94S HLSEARCH
「HLSEARCH」のキーワードを追加することで、ホスト−リガンド配位探索が行われます。
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
acetic_acid_dimer.molとacetic_acid_dimer.iniをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par acetic_acid_dimerenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
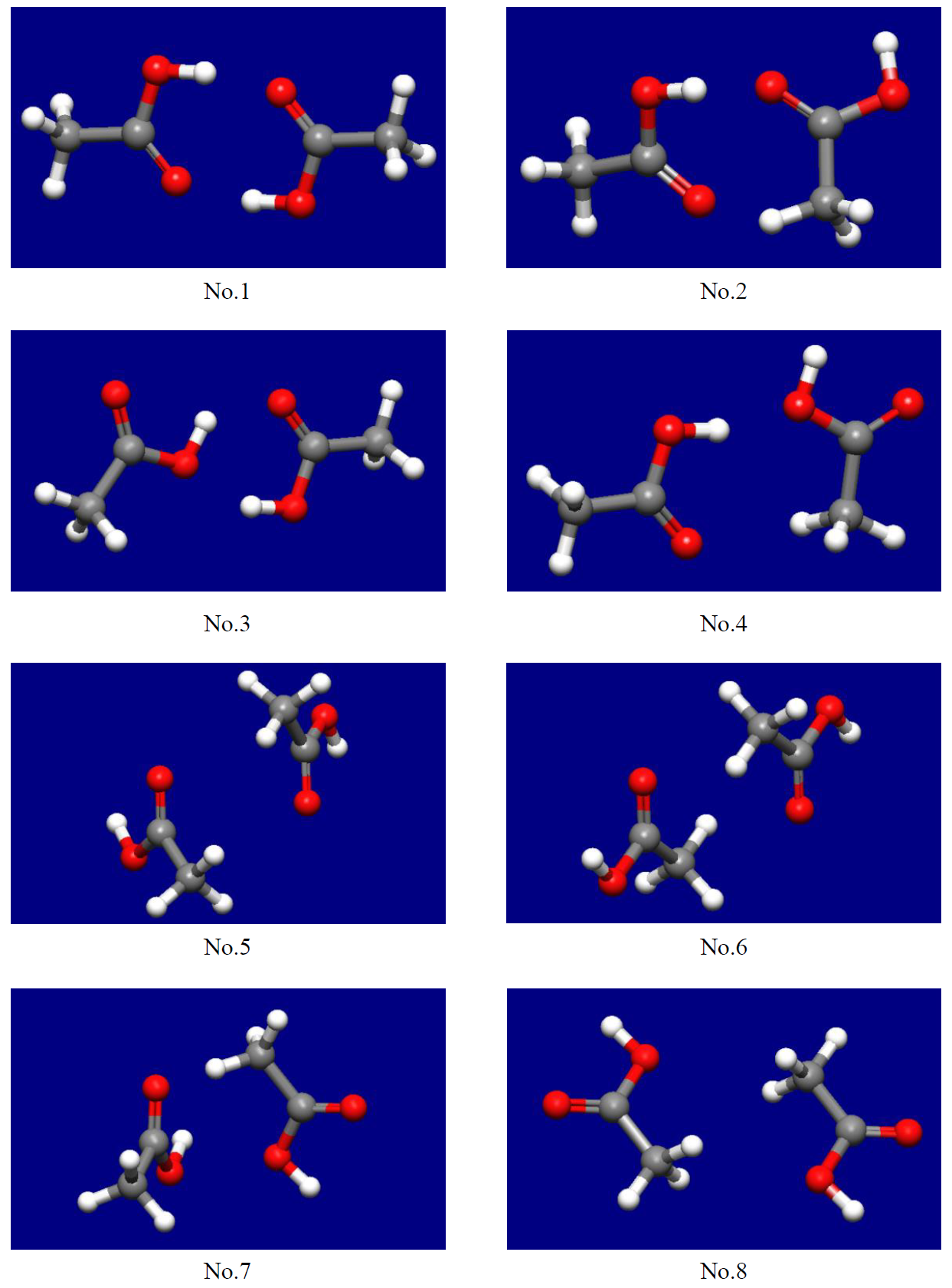
ホスト−リガンド配位探索の結果、8通りの二量体構造が得られます(下図)。
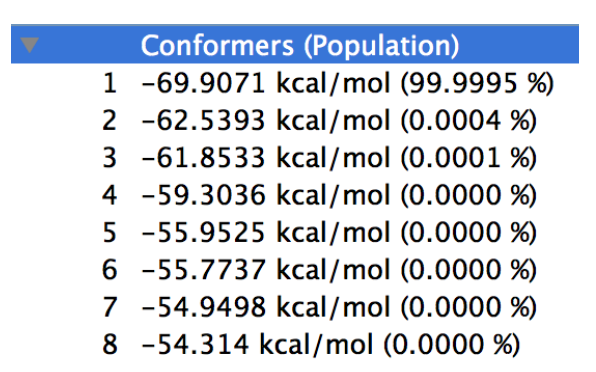
またエネルギー値のリストは、配座探索計算と同様Property Boxに表示されます(下図)。
可視化の方法は、「計算結果の可視化」をご覧ください。
【グルコース分子への水分子の配位探索】
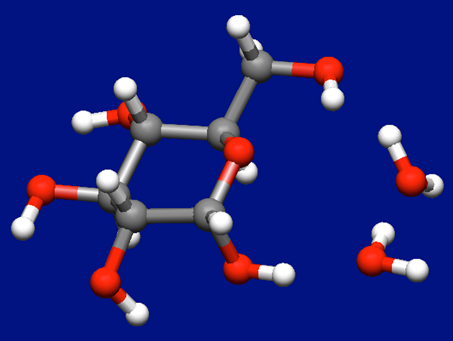
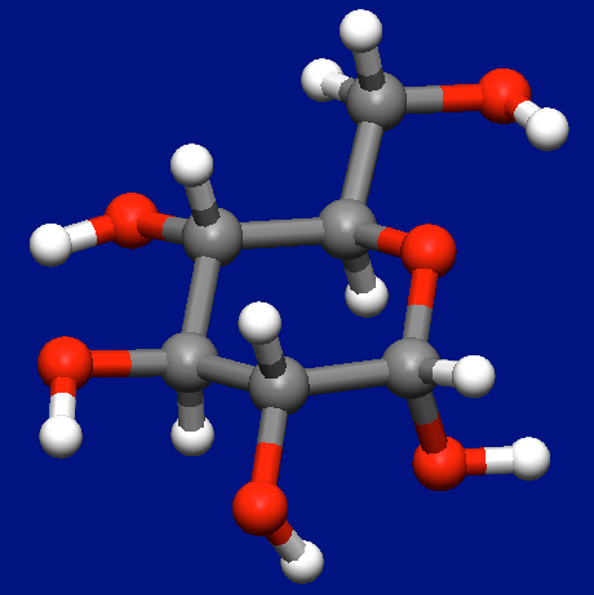
α-D-グルコース分子(以下グルコース)に水分子を1つおよび2つ配位した構造を探索します。
最初にグルコース単体での配座探索を行い最安定構造を求めました(下図)
そこに水分子を1つ配位した入力構造を作成しました(下図)。
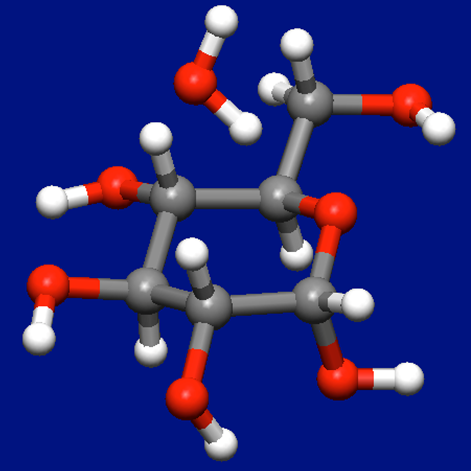
α-D-グルコース+水分子の入力構造データ(aDglucose_H2O.mol)
aDglucose_H2O.mol
27 26 0 0 0 999 V2000
-0.5024 2.4148 0.6016 O 0 0 0 0 0
-0.8520 -1.0932 -0.5731 O 0 0 0 0 0
0.4519 -1.5744 -0.2774 C 0 0 0 0 0
-0.2020 1.2411 -0.1553 C 0 0 0 0 0
-1.1949 0.1125 0.1452 C 0 0 0 0 0
2.1431 1.8092 -0.2200 O 0 0 0 0 0
0.5330 -1.9764 1.0847 O 0 0 0 0 0
2.8319 -0.9820 -0.1321 O 0 0 0 0 0
-2.6186 0.5024 -0.2701 C 0 0 0 0 0
1.5391 -0.5233 -0.5592 C 0 0 0 0 0
1.2184 0.7799 0.1723 C 0 0 0 0 0
-3.5094 -0.5881 -0.0183 O 0 0 0 0 0
0.2794 2.9953 0.4978 H 0 0 0 0 0
0.6445 -2.4621 -0.8896 H 0 0 0 0 0
-0.2535 1.5201 -1.2154 H 0 0 0 0 0
-1.2141 -0.1177 1.2184 H 0 0 0 0 0
3.0267 1.3889 -0.1951 H 0 0 0 0 0
-0.0183 -2.7768 1.1469 H 0 0 0 0 0
2.6839 -1.4193 0.7318 H 0 0 0 0 0
-2.6702 0.7196 -1.3421 H 0 0 0 0 0
-2.9791 1.3731 0.2851 H 0 0 0 0 0
1.6027 -0.3262 -1.6358 H 0 0 0 0 0
1.3472 0.6668 1.2558 H 0 0 0 0 0
-3.0727 -1.3725 -0.4004 H 0 0 0 0 0
-0.3106 0.8234 -2.7522 O 0 0 0 0 0
-0.3290 0.0085 -2.2451 H 0 0 0 0 0
-0.9697 0.7822 -3.4490 H 0 0 0 0 0
1 4 1 0 0 0 0
1 13 1 0 0 0 0
2 3 1 0 0 0 0
2 5 1 0 0 0 0
3 7 1 0 0 0 0
3 10 1 0 0 0 0
3 14 1 0 0 0 0
4 5 1 0 0 0 0
4 11 1 0 0 0 0
4 15 1 0 0 0 0
5 9 1 0 0 0 0
5 16 1 0 0 0 0
6 11 1 0 0 0 0
6 17 1 0 0 0 0
7 18 1 0 0 0 0
8 10 1 0 0 0 0
8 19 1 0 0 0 0
9 12 1 0 0 0 0
9 20 1 0 0 0 0
9 21 1 0 0 0 0
10 11 1 0 0 0 0
10 22 1 0 0 0 0
11 23 1 0 0 0 0
12 24 1 0 0 0 0
25 26 1 0 0 0 0
25 27 1 0 0 0 0
M END
[Interfaceから実行する場合]
aDglucose_H2O.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
詳細設定ダイアログが開きます。
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」 のプルダウンメニューから「Host Ligand Search」を選択します。
設定が終わりましたら、詳細設定ダイアログのをクリックします。
開いたダイアログに、「OPT=GROUP」と「MOL_GROUP=(25,1)」を追加します。
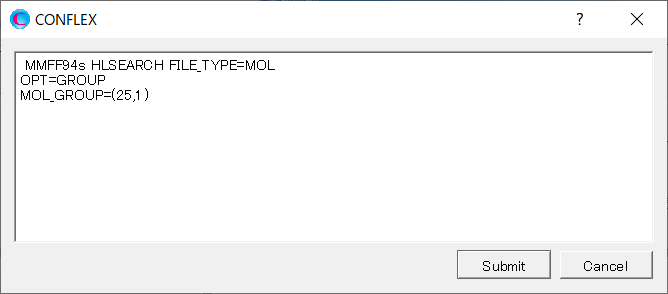
ここで「OPT=GROUP」および「MOL_GROUP=(25,1)」のキーワードを追加したことで、グルコースの構造は固定したままで水分子の位置と構造のみ最適化を行います。
部分固定に関しては、「構造を部分的に固定した計算 → 分子単位での構造固定」をご覧ください。
設定が終わりましたら、をクリックします。ホスト−リガンド配位探索が行われます。
[コマンドラインから実行する場合]
計算設定は、aDglucose_H2O.iniファイルにキーワードを記述することで行います。
aDglucose_H2O.iniファイル
HLSEARCH OPT=GROUP MOL_GROUP=(25,1)
「HLSEARCH」のキーワードを追加することで、ホスト−リガンド配位探索が行われます。
また、「OPT=GROUP」および「MOL_GROUP=(25,1)」のキーワードを追加したことで、グルコースの構造は固定したままで水分子の位置と構造のみ最適化を行います。
部分固定に関しては、「構造を部分的に固定した計算 → 分子単位での構造固定」をご覧ください。
aDglucose_H2O.molとaDglucose_H2O.iniをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par aDglucose_H2Oenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
上記の計算により、10通りの構造が得られました。最安定構造を下図に示します。
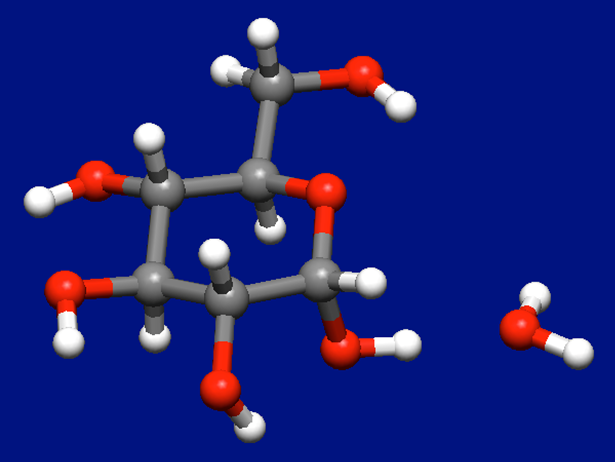
水分子を2つ含む場合
上図の構造に対して水分子をもう一つ加えて以下の構造を作り、再度配置探索計算を行います。
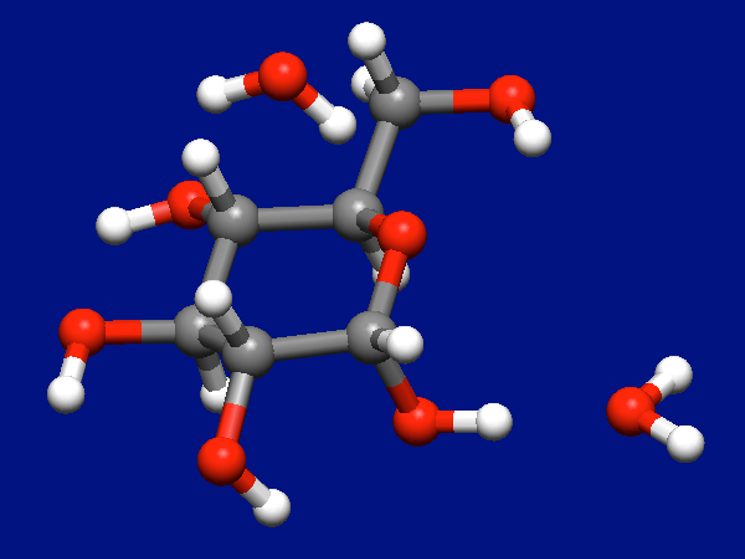
α-D-グルコース+水2分子の入力構造データ
aDglucose_2H2O.mol
30 28 0 0 999 V2000
-1.7903 2.0810 -0.8582 O 0 0 0 0 0
0.9497 0.1820 0.7911 O 0 0 0 0 0
0.4899 -1.1464 0.5531 C 0 0 0 0 0
-1.1899 1.1508 0.0453 C 0 0 0 0 0
0.3377 1.1585 -0.0836 C 0 0 0 0 0
-3.1447 -0.2633 -0.0402 O 0 0 0 0 0
0.9130 -1.5822 -0.7345 O 0 0 0 0 0
-1.4750 -2.5911 0.2513 O 0 0 0 0 0
0.9177 2.5280 0.2908 C 0 0 0 0 0
-1.0411 -1.2797 0.6520 C 0 0 0 0 0
-1.7220 -0.2507 -0.2493 C 0 0 0 0 0
2.3453 2.4922 0.1999 O 0 0 0 0 0
-2.7454 1.8632 -0.8498 H 0 0 0 0 0
0.9541 -1.8120 1.2891 H 0 0 0 0 0
-1.4901 1.4462 1.0585 H 0 0 0 0 0
0.6456 0.9385 -1.1142 H 0 0 0 0 0
-3.3916 -1.2089 0.0143 H 0 0 0 0 0
1.8682 -1.7501 -0.6492 H 0 0 0 0 0
-0.9383 -2.8164 -0.5360 H 0 0 0 0 0
0.6697 2.7883 1.3250 H 0 0 0 0 0
0.5535 3.3193 -0.3704 H 0 0 0 0 0
-1.3717 -1.1301 1.6858 H 0 0 0 0 0
-1.5810 -0.5013 -1.3080 H 0 0 0 0 0
2.6160 1.6280 0.5609 H 0 0 0 0 0
3.5948 -1.8804 -0.5097 O 0 0 0 0 0
4.1240 -1.1431 -0.8632 H 0 0 0 0 0
4.2686 -2.5619 -0.3423 H 0 0 0 0 0
-0.2472 0.9616 3.3577 O 0 0 0 0 0
0.3575 0.6108 2.6999 H 0 0 0 0 0
-1.1224 1.0508 2.9734 H 0 0 0 0 0
1 4 1 0 0
1 13 1 0 0
2 3 1 0 0
2 5 1 0 0
3 7 1 0 0
3 10 1 0 0
3 14 1 0 0
4 5 1 0 0
4 11 1 0 0
4 15 1 0 0
5 9 1 0 0
5 16 1 0 0
6 11 1 0 0
6 17 1 0 0
7 18 1 0 0
8 10 1 0 0
8 19 1 0 0
9 12 1 0 0
9 20 1 0 0
9 21 1 0 0
10 11 1 0 0
10 22 1 0 0
11 23 1 0 0
12 24 1 0 0
25 26 1 0 0
25 27 1 0 0
28 29 1 0 0
28 30 1 0 0
M END
[Interfaceから実行する場合]
aDglucose_2H2O.mol.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
詳細設定ダイアログが表示されます。
次に、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」 のプルダウンメニューから「Host Ligand Search」を選択します。
続いて、詳細設定ダイアログの「Host Ligand Search」ダイアログで、ホスト−リガンド配位探索計算の設定を行います。
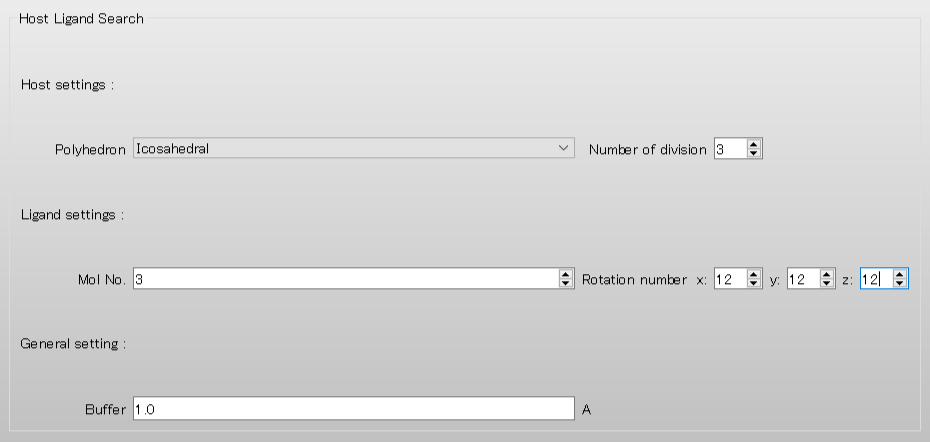
まず、「Number of division」を「3」とします。これによりリガンドとなる分子の配置場所をより細かく設定することができます。
また、「Ligand settings:」の「Mol No.」を「3」とすることで、追加した2つ目の水分子をリガンドとして指定します。
さらに、Rotation number x:, y:, z:のそれぞれの値を「12」とすることで、リガンドに対して30度刻みの分子回転が行われるようになります。
設定が終わりましたら、詳細設定ダイアログのをクリックします。
起動したダイアログに、「OPT=GROUP」、「MOL_GROUP=(25,1)」、「MOL_GROUP=(28,1)」を追加します。
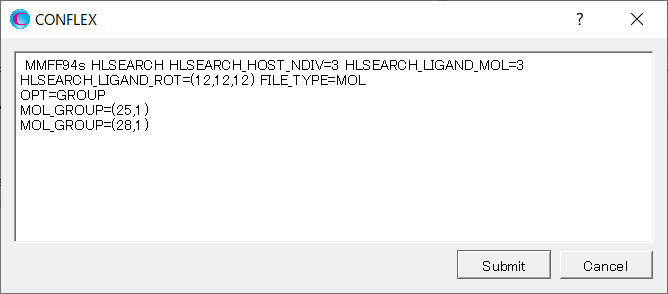
ここで「OPT=GROUP」および「MOL_GROUP=(25,1)」「MOL_GROUP=(28,1)」のキーワードを追加したことで、グルコースの構造は固定したままで水分子の位置と構造のみ最適化を行います。
部分固定に関しては、「構造を部分的に固定した計算 → 分子単位での構造固定」をご覧ください。
設定が終わりましたら、をクリックします。ホスト−リガンド配位探索が行われます。
[コマンドラインから実行する場合]
計算設定は、aDglucose_2H2O.iniファイルにキーワードを記述することで行います。
aDglucose_2H2O.iniファイル
HLSEARCH OPT=GROUP MOL_GROUP=(25,1) MOL_GROUP=(28,1) HLSEARCH_HOST_NDIV=3 HLSEARCH_LIGAND_ROT=(12,12,12) HLSEARCH_LIGAND_MOL=3
それぞれのキーワードの意味は、次の通りです。
| キーワード | 説明 |
|---|---|
| HLSEARCH | ホスト−リガンド配位探索が行われます。 |
|
OPT=GROUP MOL_GROUP=(25,1) MOL_GROUP=(28,1) |
グルコースの構造は固定したままで水分子の位置と構造のみ最適化を行います。 |
| HLSEARCH_HOST_NDIV=3 | リガンドとなる分子の配置場所をより細かく設定します |
| HLSEARCH_LIGAND_ROT=(12,12,12) | リガンドに対して30度刻みの分子回転が行われるようになります。 |
| HLSEARCH_LIGAND_MOL=3 | 追加した2つ目の水分子をリガンドとして指定します。 |
aDglucose_2H2O.molとaDglucose_2H2O.iniをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par aDglucose_2H2Oenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
上記の設定で得られた最安定構造を図に示します。