Docking simulation flow
This section describes the flow of docking simulation using CONFLEX DOCK.
CONFLEX DOCK is a docking simulation program that predicts where a specified peptide chain will coordinate with a substrate protein to form a complex.
When making predictions, the amino acid residues of the protein are first coarse-grained at representative points (Cα atoms) and a tetrahedron is constructed by Delaunay partitioning. Then, peptide residues are placed on search points set on the protein surface, and scores are evaluated based on the tetrahedral coarse-grained potentials.
[Literature cited]
T. Yamamoto, Y. Ikabata, H. Goto, “Reconstruction of Four-Body Statistical Pseudopotential for Protein-Peptide Docking”, J. Comput. Chem., Jpn.-Int. Ed., 2024, 10, 2023-0039
In CONFLEX DOCK, the program that performs the calculation and the Interface run separately, so calculations can be performed with or without the Interface. We will first explain how to run a calculation from the Interface, and then show you how to run a calculation by entering a command.
CONFLEX Interface does not have the ability to create molecules, so existing PDB format files must be opened and used.
Open PDB file
Select “Open” from the “File” menu to display a dialog box.
In the “Files of type” pop-up at the bottom of the dialog, select “PDB File (*.pdb)” as the format of the file to open, then open the desired PDB file.

Select the desired file and press the “Open” button to open the file and display it in the graphic window.
Start of docking calculation
To start the calculation, select “Docking” from the “Calculation” menu. Once selected, the following dialog will appear:
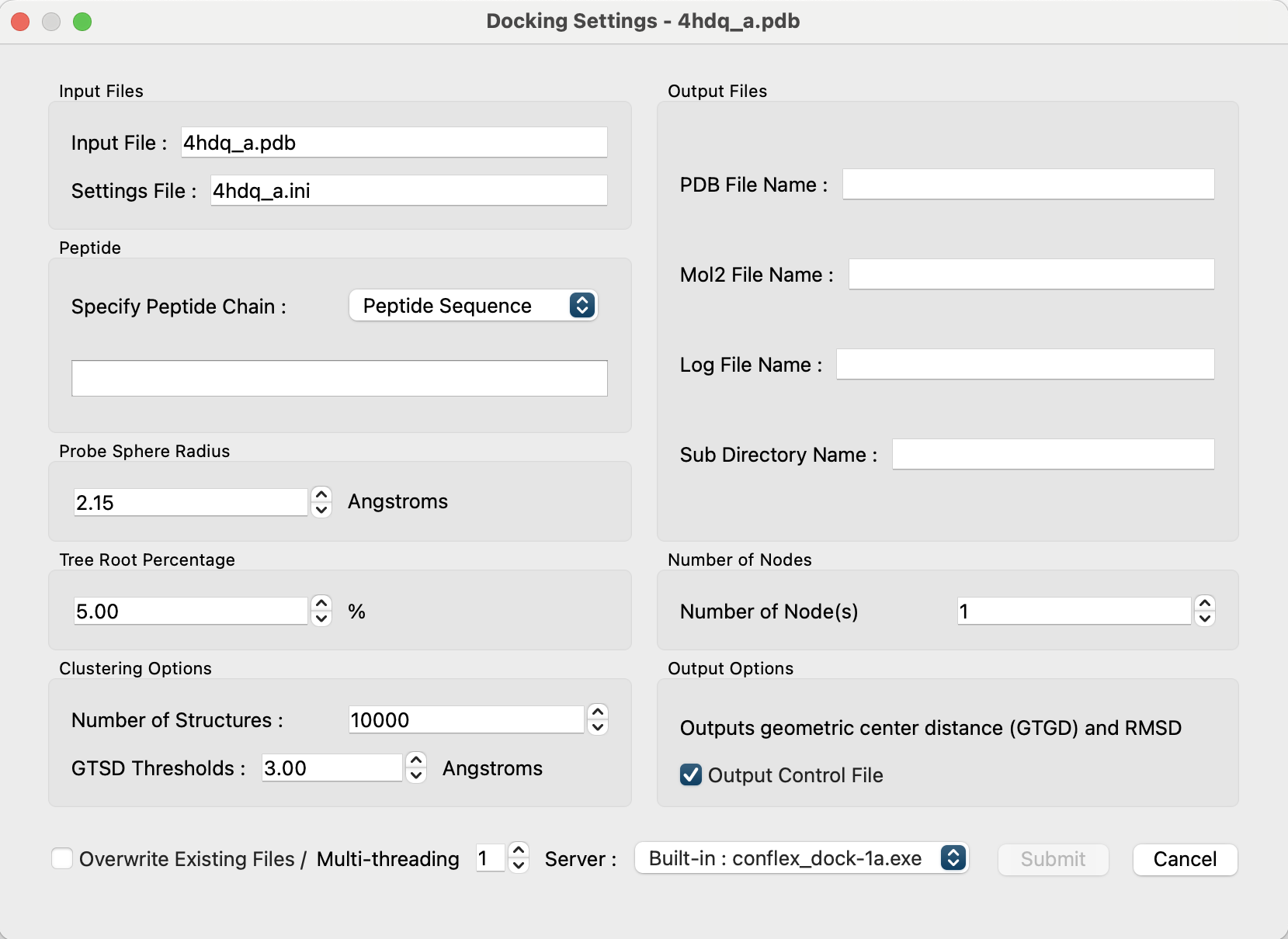
The contents of the dialog are described below:
- Input Files:
-
The name of the input PDB file to be used for the calculation and the name of the input file where the calculation settings will be saved are indicated.
- “Input File:” shows the name of the PDB file that was opened. Editing is not allowed.
-
The “Settings File:” will store the settings made in this dialog.
A default name is shown with the molecular filename extension changed from pdb to ini. It is possible to edit it and change it to another name.
- Peptide
-
Specifies the peptide to be docked.
Under “Specify Peptide Chain:” select how you want to specify the peptide.-
If “Peptide Sequence” is selected, enter the peptide sequence in the box below.
Both 1- and 3-letter codes are recognized. 3-letter codes must have “-” between the amino acids.
Example: ARG-ARG-ASP-TYR-PHE -
If “Open File” is selected, the “Open File” button appears.
Clicking on it opens a file dialog that allows you to open a separately prepared PDB file of the peptide.
-
If “Peptide Sequence” is selected, enter the peptide sequence in the box below.
- Output Files
-
Specifies the name of the file and directory output by the docking calculation. The default is the open protein PDB filename body + "_" + peptide chain notation + extension.
- “PDB File Name:” A series of pose structure will be saved after docking.
- “Mol2 File Name:” A series of representative point structure will be saved after docking.
- “Log File Name:” Logs of docking calculation will be saved.
- “Sub Directory Name:” The name of the directory where multiple data files will be stored during docking calculation.
- Probe Sphere Radius
- Sets the probe sphere radius used to locate the search points.
- Default is 2.15Å
- Tree Root Percentage
- After placing a residue at a search point and evaluating the score, the percentage of points that will be adopted as the root of the tree structure and proceed with the search is specified.
- Default is 5%
- Number of Nodes
- Select nodes to proceed with the search by number of nodes from the order of highest to lowest score.
- Default is 1.
- Clustering Options
- Under “Number of Structures”, specify the number of top-scoring predicted structures to be considered for clustering (default is 10,000).
- Under “GTSD Thresholds,” specify the GTGD thresholds for the two structures that will be used as a measure of clustering (default is 3.0Å).
- Output options
- The “Output Control File checkbox” controls the output of Geometric center To Geometric center Distance (GTGD) and Root Mean Square Deviation (RMSD).
- Valid only if the peptide chain is specified in the PDB file (default: true)
- Overwrite Existing Files
- If the check box is checked, the specified output files will be overwritten even if it exists.
- Default is off.
- Multi-threading
- Specify the number of parallel operations by OpenMP.
- Default is 1.
- Server:
- Specify the executable file to perform the calculation. Basic settings are made in ”Preferences...”.
- Submit button
- Click to submit the calculation job.
- The start of the calculation is determined by the current job status.
Job Manager
When the “Submit” button in the docking dialog is clicked, the job is not started immediately. The job is registered in the Job Manager. If the CPU of the specified computer is free, the job will be started. The “Job Manager” dialog appears automatically when a job is submitted. To display it manually, select “Job Manager” from the “Tools” menu.
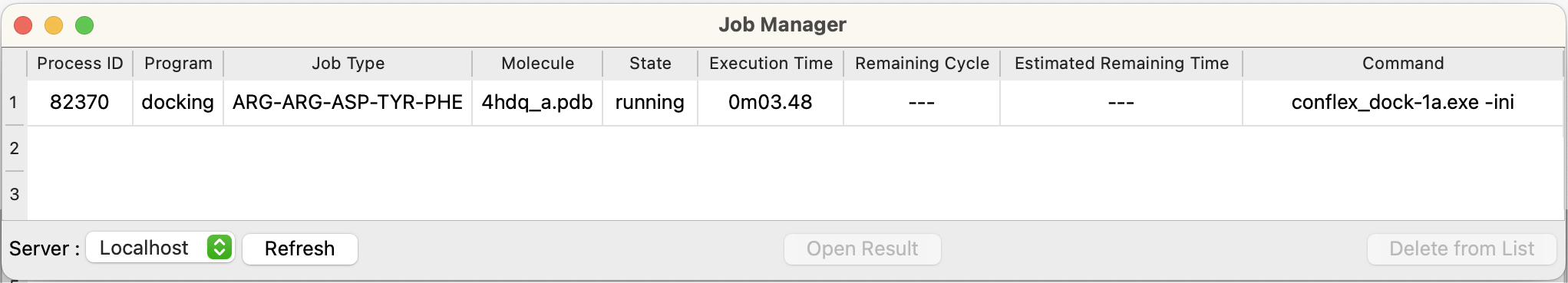
The job's progress will be checked every number of seconds specified in the “Preferences”, and the dialog display will be updated. If you have not specified an automatic check, select the server you want to display from the popup in the lower left corner of the dialog and click the “Refresh” button. “Server:Localhost” means the computer currently running Interface. Each item consists of the following contents.
- Process ID
- The identification number of the program being executed.
- Program
- The name of the executable program is displayed.
- Job Type
- The sequence of peptides that are being docked is displayed.
- Molecule
- The file name of the molecule being calculated is displayed.
- State
- The program execution status is displayed. When the program is submitted, calculation marked as “pending”. When execution starts, the status will be “running† and when it finishes, “Finished” will be displayed.
- Execution Time
- The elapsed time since the start of execution is displayed.
- Remaining Cycle
- Not shown for docking.
- Estimated Remaining Time
- Not shown for docking.
- Command
- Indicates the command being executed.
Display of calculation results
Jobs that are “Finished” in the “Job Manager” can be observed by opening the calculation results.
After clicking on a job labeled “Finished” to select it, click the “Open Result” button at the bottom center of the dialog or double-click the list item to open the calculation results.
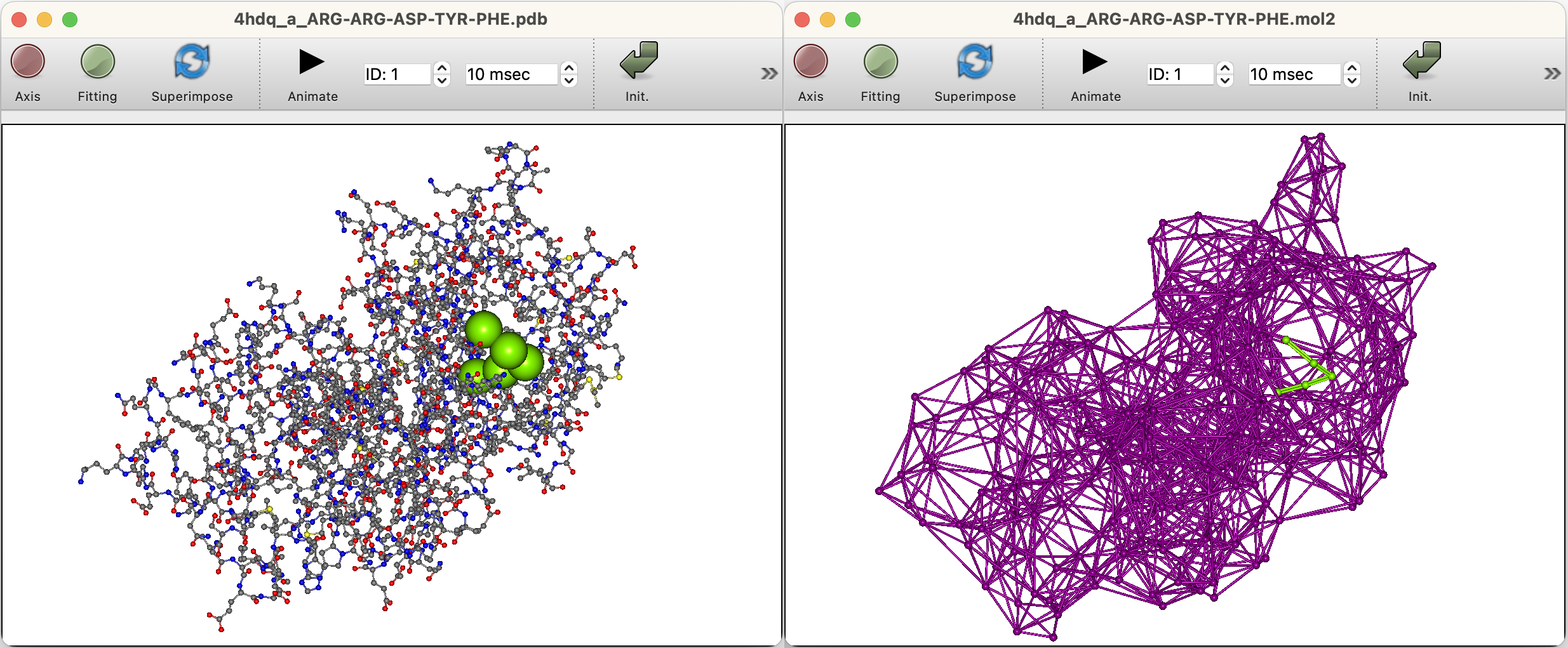
For docking calculations, the PDB output file specified in the calculation setup screen will be opened.
If you want to open a Mol2 file in which representative points have been saved, select “Open...” from the “File” menu. Then, specify the file type “Sybyl Mol2 Files (*.mol2)” in the dialog box and open the mol2 file saved.
Docking pose list
When the file resulting from the docking calculation is opened, the structure with the highest score is displayed in the graphic window, and a list of docking scores appears in the “Property Box”. Clicking on an item in the list to select it will change the structure in the graphic window to that docking pose.
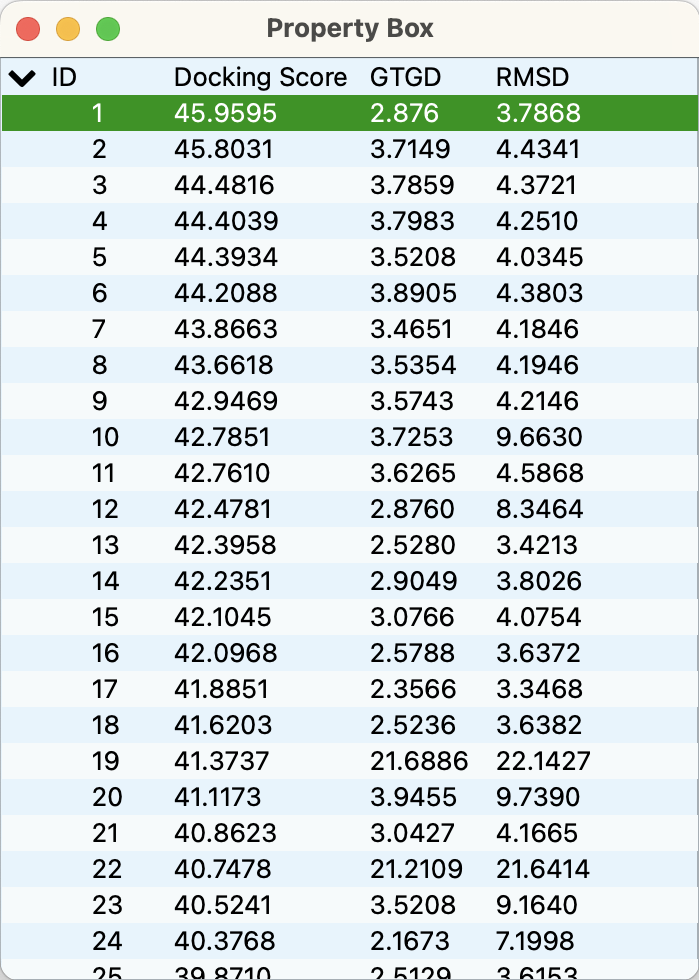
Multiple structures can be selected by holding down the Shift key and clicking in the “Property Box” to display them overlapping in the molecular screen.