- 製品&サービス
- CONFLEX
CONFLEXは、フレキシブルな分子の配座空間を探索し、化学的に重要な配座異性体の最適化構造をもれなく見つけだします。今までの構造最適化プログラムでは、ユーザーが入力した初期構造に依存した、局所的な最適化構造しか求めることができませんでした。
実践的に意味のある安定な配座異性体を優先的に創出することにより、効率的な配座空間探索を実現します。CONFLEXは、材料開発、創薬研究、有機合成、分子設計などの分野で幅広く利用されています。
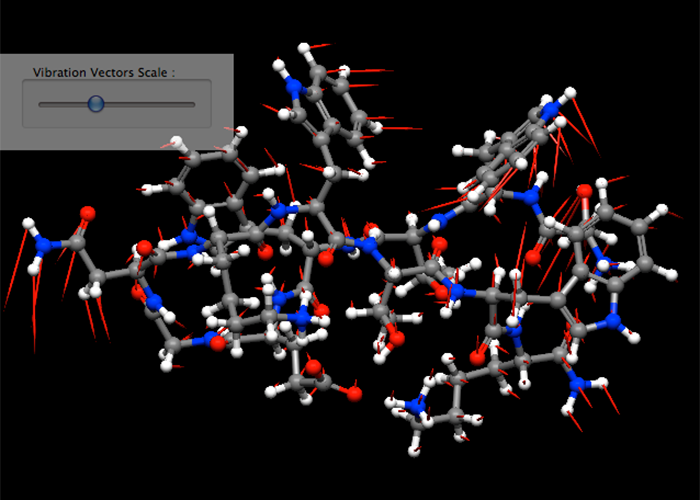

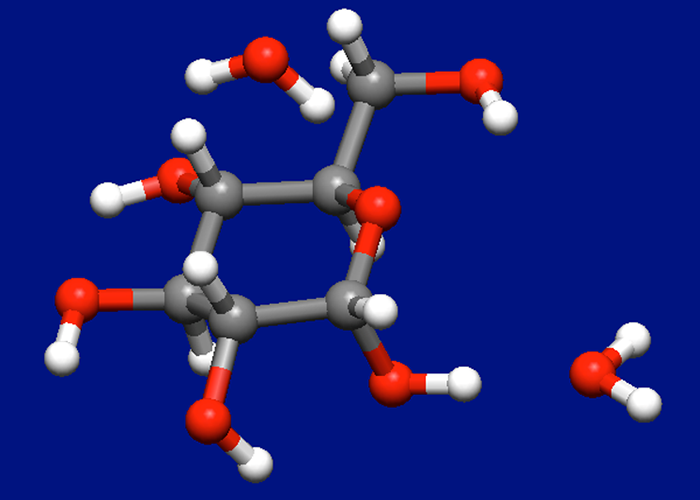
配座空間探索
分子内の環と直鎖部分を自動的に判別し、環にはCorner FlapおよびEdge Flip、直鎖にはStepwise Rotationを行うことで初期構造を創出します。それら全てに対して構造最適化を行い得られた配座異性体を保存します。この際、貯水池注水アルゴリズムにより、エネルギー的に安定な配座から常に初期構造を創出します。さらに探索範囲を、最安定構造からのエネルギー差により指定できます。
これらにより、最安定構造探索の効率化と探索配座数の爆発的な増加を防ぐことができます。
基準振動解析
構造最適化で得られた極小構造に対して、自動的に基準振動解析を行います。
基準振動解析により得られた振動モードの表示、およびGibbsの自由エネルギーなどの熱力学的諸量を算出します。
構造を部分的に固定して最適化することも可能です。
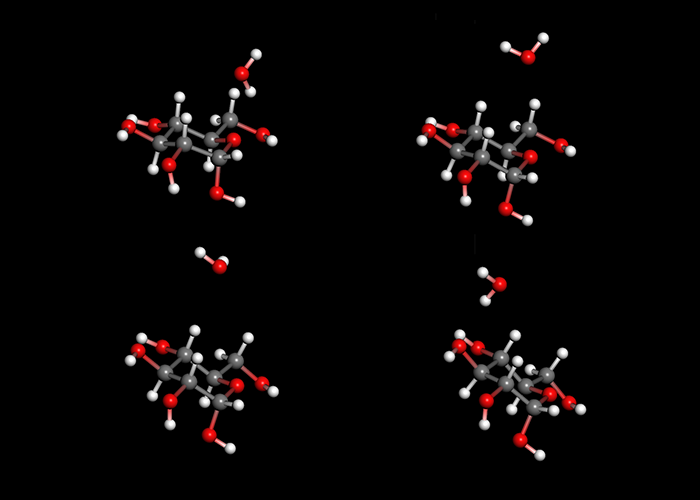
分子性結晶&結晶構造探索
分子構造データと空間群の対称性を入力することで、自動的に結晶構造を作成して構造最適化を行ない、エネルギー極小に位置する結晶構造を網羅的に算出します。
最適化した一連の結晶構造に対して、エネルギーの低い順に並べるだけでなく、あらかじめ用意した粉末回折データに近い順に並べることもできます。
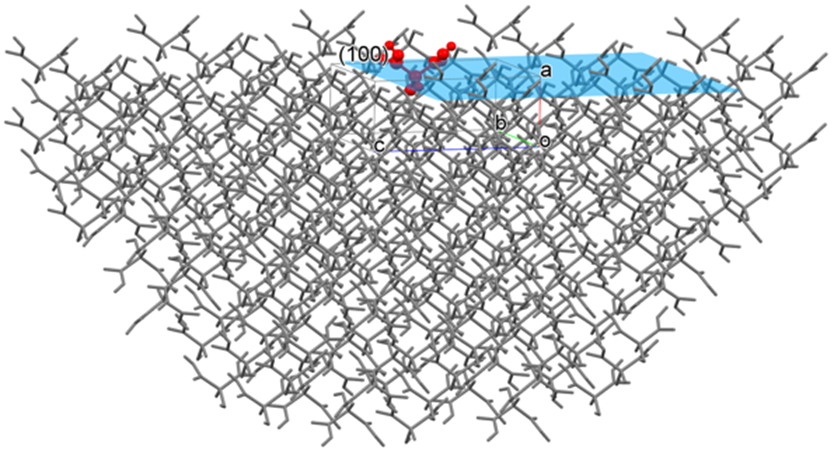
結晶表面解析
結晶表面の解析は、結晶の溶解や昇華など、結晶表面で起こる現象を理解する上で重要です。
CONFLEXは、指定した結晶面における分子の相互作用エネルギーを求め、その結晶面の安定性等を評価することができます。例えば、(100)面を指定した場合、CONFLEXは下図に示すように、(100)面を露出させた半球結晶を構築し、(100)面を構成する分子、あるいはその結晶面上に存在する分子の分子間相互作用エネルギーの和を求めることができます。なお、半球の半径は設定したカットオフ距離になります。
ここで、Emolは露出させた結晶面の中心に存在する分子(上図、赤色の分子。以下、基本分子と呼ぶ)の分子間相互作用エネルギーの和であり、Nは基本分子の総原子数、Mは基本分子以外の半球結晶に含まれる全分子の総原子数です。

溶媒効果
GB/SAモデルを用いた計算が、構造最適化、振動解析および、配座解析で可能です。
また、LogP値を自動的に算出することが出来ます。
Gaussian連携機能
同じマシンにGaussianがインストールされている場合、CONFLEXからGaussianを呼び出して、構造最適化および配座探索計算を直接行うことができます。
これにより、分子力場パラメーターの無い分子や、古典力場では扱えない電子状態での構造最適化・配座探索が可能となりました。
Dynamic Reaction Coordinate (DRC)
Dynamic Reaction Coordinate (DRC) は、基準振動モードを使用して初期速度ベクトルを算出し、動力学計算を行う計算方法です。
複数分子の配置変換や、大きな分子の配座変換に適用できます。
ホストーリガンド配位探索
複数の分子を含む系について、ある一つの分子が他の分子(群)に対して どの位置にどのような向きで配位するのが安定かを探索します。 二量体や錯体の安定構造の探索に利用出来ます。
その場配向探索
複数の分子を含む系で、特定の分子を入力構造の位置で回転させ、安定な配向を探索する機能です。
CD/UV/Visスペクトル解析
CONFLEXで得られた配座異性体についてCD/UV/Visスペクトル計算を行うことが可能です。
NMR結合定数解析
有機化合物の構造を解析する上で、NMR解析は欠くことのできない測定法の一つです。 特に、ビシナル位(1,4位)にある原子同士の結合定数3Jは、2,3位の原子間の結合を回転軸とした時の二面角に依存することから、適切な数式とパラメーターを設定することで有機化合物の立体構造を推定することができます。最近では、この結合定数に基づく二面角と、NMR-NOE情報による近接プロトン間距離の予測値を組み合わせることによって、タンパク質など生体高分子の立体構造を予測することも可能になっています。一方で、分子が大きくなるにつれ考慮すべき配座異性体の数も増加するため、NMR解析で得られる結合定数などの測定値は配座異性体混合物の平均値として観測されます。このため、測定値だけを頼りにしてしまうと、現実には存在しないような「不自然な」平均立体構造が予測される可能性が高まります。 CONFLEXでは、
- Karplus-Imai式を用いたCsp3-Csp3結合周りの3JHHの算出
- ユーザーが指定したパラメーターセットを用いたKarplus式による3Jの算出
の2通りの計算を行うことが可能です。さらに、配座探索によって創出された個々の配座異性体に対して3J値を計算し、存在比に基づく熱力学平均値を求めることもできます。こうして得られた計算値を測定値と比較することによって、NMR解析で観測されている分子の状態を推定し、物性や反応性に及ぼす立体効果を予測するなど様々な立体配座解析や、より正確な立体構造の解析を支援することができます。
パラメーター設定
CONFLEXに含まれている力場パラメーターに対応していない原子タイプを含む分子について、パラメーターを追加して計算することができます。
既存のパラメーターを修正して計算することも可能です。MMFF94s力場のみの対応です。
アミノ酸残基置換機能
生体内に存在するタンパク質は何らかの影響でいくつかのアミノ酸残基が変異することがあります。数残基しか変異していないタンパク質でも、その機能や活性が劇的に変化する場合があります。そのような現象を計算機で調べるためには、あらかじめ用意されているアミノ酸配列が記述されているPDBフォーマットを使用し、それに対してアミノ酸の置換を行なうのがよいでしょう。
また、PDBフォーマットを使用する場合、多くの構造はX線結晶解析実験で得られており、運動の大きいアミノ酸側鎖の一部の構造が決定されていない例があるため、不足分子を補う必要があります。
CONFLEXでは、PDBファイルに対してこれらのアミノ酸側鎖の置換や補足を行ない、その後の計算を行なうことができます。また、アミノ酸側鎖の不足情報のほかにも、主鎖の不足情報を得ることができます。