分子の構築
この章では、GaussViewでの分子構築の機能について説明します。ここで示すオプションにより、原子ごとあるいはフラグメントを用いた新規分子構造の構築だけでなく、すでに計算した分子の操作や解析が可能となります。GaussViewの表示機能を用いるあるいは分子ファイルや画像を開いたり保存する際の詳細については、このマニュアルの「分子ファイルと画像の操作」をご覧ください。
元素とフラグメントの配置
GaussViewの分子構築ツールは常にアクティブビューウィンドウ上で動作します。通常、以下に示すオプションを異なる方法で実行するよう明示的に呼び出されなければ、GaussViewは挿入/置換モードで開始しかつその状態を保ちます。ビューウィンドウの空きスペースをクリックすると、現在選択されている原子あるいはフラグメントが独立したフラグメントとしてウィンドウに追加されます。ウィンドウ内に存在する原子上をクリックすると、現在選択されている項目が分子構造中の指し示したところに、通常は置き換えられるかたちで追加されます(他のオプションについては後述します)。
希望する原子やフラグメントは、Element, Functional Groups, Rings , Biological Fragmentsボタンの中の1つを用いて、現れたパレットにある選択肢から1つ選ぶことで選択します。現在選択されている項目は、GaussView コントロールウィンドウのカレントフラグメントエリア内および/またはスタンドアローンのビルダーパレットにあるアクティブフラグメントエリア内(図3参照)に表示されます。スタンドアローンのビルダーパレットにあるアクティブフラグメントエリアあるいはメインウィンドウのカレントフラグメントエリアの上にあるフラグメント名をクリックすると、選択されているフラグメントタイプに対応したパレットが開きます。
スタンドアローンのビルダーパレットはView=>Builder メニュー項目を選択することで開くことと、Window Behavior環境設定(File=>Preferencesメニューでアクセスする)にあるBuilder Dialog Stays on Topチェックボックスをチェックすることで、開いている全てのウィンドウの最上位に置くようにすることが出来ます。
Elementパレット
Element Fragmentsウィンドウでは、周期表から元素を選ぶことが可能です。選択した元素に対して可能な各種の配位形式がウィンドウの下部に表示されます(図10参照)。デフォルトでは4面体型炭素原子が表示されます。
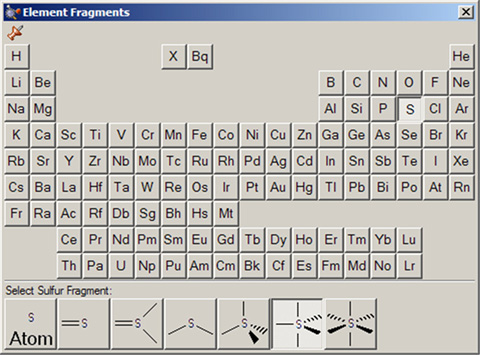
図10.選択されたElementパレット
選択した元素に対して利用できる混成(結合)の種類がパレットの下部に沿って現れている。
この場合、選択されている原子タイプは5価の硫黄原子である。
Groupパレット
Group Fragmentsウィンドウでは、AM1で最適化された一連の官能基フラグメントを選ぶことが可能です(図11)。デフォルトではカルボニル基(ホルムアルデヒド)が表示されます。
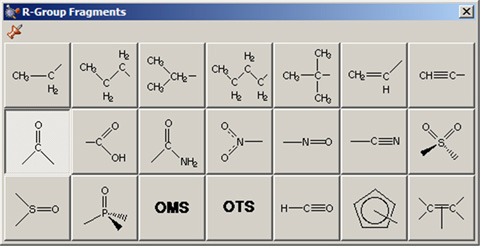
図11.R-Group Fragments選択パレット
希望する官能基を選択するのにこのパレットを用います。
Ringsパレット
Ring Fragmentsウィンドウでは、AM1で最適化された一連の環構造を選ぶことが可能です(図12)。デフォルトではベンゼン環が選択されています。

図12.Ring Fragments選択ウィンドウ
希望する環系を選択するのにこのパレットを用います。
フラグメントのいくつかは結合状況が不自然になっていることに注意してください。それらには、環平面に垂直な方向に開いた結合を持つ5員環が含まれています。これは有機金属系の構築に用いられるシクロペンタジエニル配位子です。
Bioパレット
Biological Fragmentsダイアログでは、AM1で最適化された生物学の研究者にとって有用なフラグメントであるアミノ酸や様々なDNA塩基(図13)を提示しています。デフォルトでは中心残基のアラニンが表示されます。
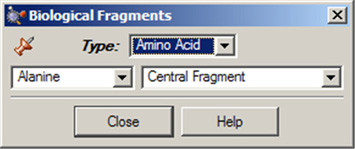
図13.Biological Fragmentsの選択
アミノ酸やDNA塩基をこのウィンドウ内の欄を使い選択します。
ダイアログのType欄ではAmino AcidかNucleosideのいずれかを選択できます。Amino Acidを選択した場合、Type欄の下にある左側のポップアップメニューでは希望するアミノ酸を選択することが出来、また同じ列にある右側のポップアップメニューでは構造様式をCentral Fragment、Amino-Terminal Fragment、もしくはCarboxyl-Terminal Fragment(後の2つはそれぞれN末端型およびC末端型です)から指定することが出来ます。
Type欄でNucleosideを設定した場合、Type欄の下にある左側のポップアップメニューでは希望するDNA塩基を選択することが出来、また同じ列にある右側のポップアップメニューでは構造様式をCentral Fragment, C3’-Terminal Fragment, C5’-Terminal Fragment もしくはFree Nucleosideから指定することが出来ます。
パレットの貼り付け
GaussViewのフラグメントパレットは、他のパレットが開くと閉じる(ダイアログボックスの挙動)モーダル様式か、あるいは明示的に閉じるまでずっと開いている(浮いているパレットの挙動)アモーダル様式のいずれかで使うことが出来ます。ウィンドウの左上の角にある押しピンアイコンは、他のパレットが開いたとき(あるいは分子構築に関係のないコンテクストを入力したとき)にそのパレットが閉じるかどうかを示しています。差し込まれたびょうは、パレットがデスクトップ上に”びょうで留められている”(すなわち”くっついている”)ことを示しています。上向きにとがったびょうも同様にモーダル操作であることを示しています。
UndoとRedo
メニューのEdit=>UndoとEdit=>Redoを選択すると、一つ前の分子の修正もしくはその他の操作を無効にしたり元に戻すために用いられます。UndoボタンとRedoボタンも、同じ目的で用いられます。
構造パラメーターの表示と調節
「Builder」ツールバー/ウィンドウは、分子の構造パラメータを表示あるいは修正するための以下のボタン(Bond, Angle, Dihedral, Inquire, Add Valence, Delete Atom, Invert About Atom)を含んでいます。加えて、他のボタンやそれらに対応するメニュー項目―Clean, Rebond, Symmetrizeを含む―も分子構造とその表示に影響を及ぼします。この節ではこれらの特徴について説明します。
Inquireモード
Inquireボタンによりモードを変更後、関心のある原子をクリックして構造情報をビューウィンドウに直接表示させることが出来ます。選択した原子は、結合している必要はありません。構造情報は、図14のようにビューウィンドウのステータスバーに表示されます。選択した原子の数により、ビューウィンドウに表示される結果は以下のようになります:
- 原子上にマウスカーソルを合わせるだけ:原子タイプと番号:
 . .
- 1原子:原子タイプと番号:
 . .
- 2原子:結合長(距離):
 . .
- 3原子:原子間の角度:
 . .
- 4原子:4-3-2-1原子の2面角:
 . .
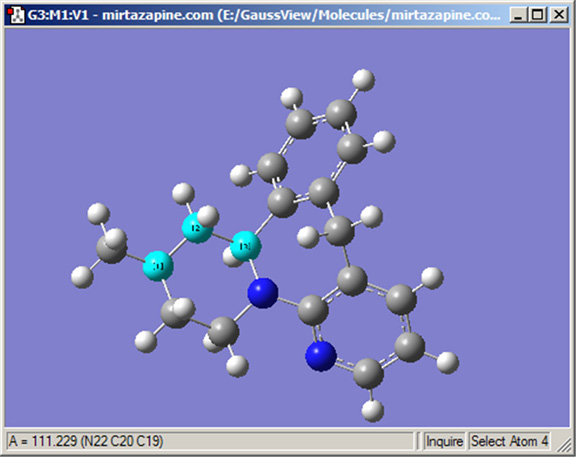
図14.Inquireモードの表示
ここではウィンドウの左下角に選択した結合角の値を表示している。
Semichem SmartSlides
Bondボタン、Angleボタン、Dihedralボタンの3つのボタンにより、構造パラメータを迅速かつ直感的に修正することが出来ます。ボタンを選択した後、それぞれのSmartSlideダイアログボックスを開くためには、対象とする原子を選択しなければなりません。SmartSlideを用いてパラメータを修正した場合、行った修正を実際に分子に適用するためにOKボタンを押す必要があります。「ウィンドウを閉じる」ボタンをクリックすると、あらゆる変更が無効になります(Cancelボタンを選択した場合と同様)。
結合SmartSlide
図15に結合のSmartSlideを示します。結合長の初期値は選択した原子対間の距離にセットされ、また原子間の結合タイプも示しています。SmartSlideでは、可能な全ての結合タイプへのアクセスを提供しています。
原子間距離は、スライダーを動かすことで動的に調節できます。数値を直接テキストボックスの中に入力することも可能です。
結合タイプラジオボタンの1つをクリックすることで、結合タイプを変更することが可能です。その際、結合が消去される原子の原子価には影響はありません。但し、これは純粋に表示上の操作であり、Gaussianで行うような量子力学的手法では接続性は考慮されず、むしろ波動関数から結合を決定することに注意してください。
結合SmartSlideは、結合していない原子間に結合を加える際にも用いられるます。従来どおり、結合させる2つの原子をクリックするだけです。SmartSlideが開いたら、希望する結合のタイプをクリックすることで、結合が作られます。同様に、結合を消去するには、2つの原子を選択し、ダイアログのNoneラジオボタンを選択します。
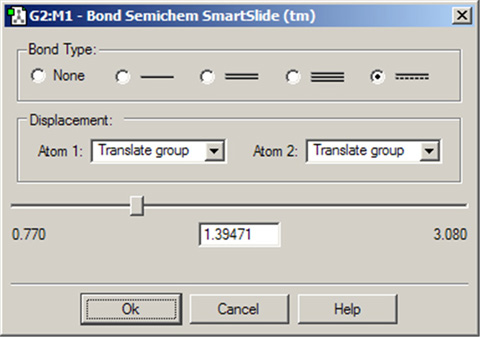
図15.結合SmartSlide
このダイアログでは、結合の追加、消去、および結合長の変更を行うことが出来ます。
Displacement欄は、結合長の変更に伴いそれらの原子に結合している原子団をどのように操作するかを特定します:
- Translate Atom:原子のみを動かし、原子団の位置は固定したままにする
- Translate Group:結合している原子団を原子の動きに沿って動かす(すなわち単一ユニットとして)
- Fixed:原子および原子団を動かさない(全ての動作は他の原子で起こる)。
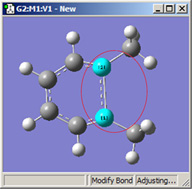
図16に、これらの選択肢の異なる組み合わせによる効果を示します。
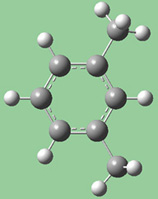 |
| 元の構造 |
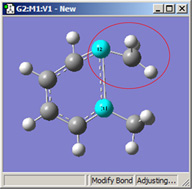 |
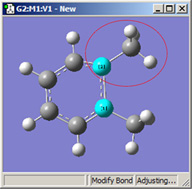 |
 |
| 1=Fixed, 2=Translate Atom |
1=Fixed, 2=Translate Group |
1,2=Translate Group |
図16.結合長修正のオプション
これら3つの構造は全て、選択した2つの炭素原子間距離を伸ばした結果です(3つ全て同じ値)。
左の絵は、原子1を固定し、原子2は Translate Atomを指定した場合です。原子1を固定し原子2にTranslate Groupを指定した、中央の絵と比べてみてください。左の場合では、上にあるメチル基は固定されています。一方中央の場合、1-2-R結合角が変わらずそのままになっています。右の絵は両方の原子にTranslate Groupを指定した場合ですが、恐らくこの場合はこれが最も一般的な選択でしょう。
結合角SmartSlideと2面角SmartSlide
図17に、結合角SmartSlideのダイアログを示します。2面角SmartSlideのダイアログは結合角SmarSlideのものと似ていますが、2面角SmartSlideでは原子1および原子4に対してのみDisplacement欄が用意されています。
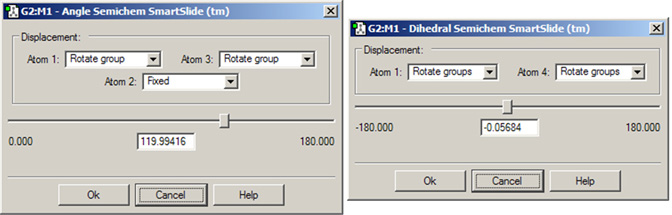
図17.結合角SmartSlideと2面角SmartSlide
結合角および2面角(選択した原子の数による)をこれらのダイアログで修正できます。
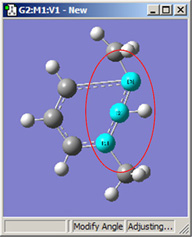
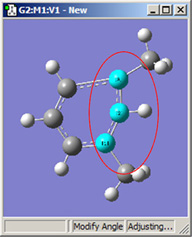
図18に、結合角についてのさまざまなDisplacementセッティングの組み合わせのいくつかを示します。(図16と同じ分子を用いています)。選べる選択肢は以下の通り:
- Rotate Atom:原子のみが動き、官能基の位置は固定したまま
- Translate Group:結合している官能基が単一ユニットとして原子の動きに沿って動く
- Rotate Group:結合角の変更に関わる原子に沿って、官能基の位置が回転する
- Fixed:原子および官能基の移動を禁止する(全ての移動は他の原子により起こる)
 |
 |
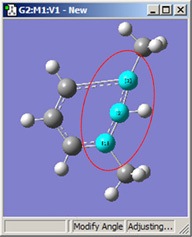 |
| 1,2=Fixed, 3=Rotate Atom |
1,2=Fixed, 3=Rotate Group |
1,2=Fixed, 3=Translate Group |
| |
|
|
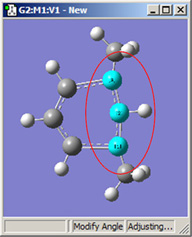 |
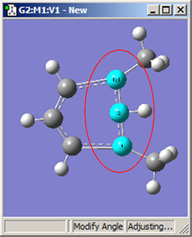 |
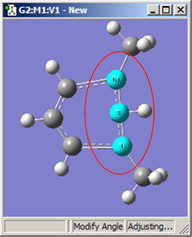 |
| 1,3=Rotate Atom, 2=Fixed |
1,3=Rotate Group, 2=Fixed |
1,3=Translate Group, 2=Fixed |
図18.結合角の修正オプション
これら6つの構造は全て、1-2-3結合角を大きくしたことで得られた構造です。
上段では、原子1と2を固定しています。結合角を同じ値だけ大きくして得られた、3つの場合での構造を比較してください(環の原子とメチル基の位置は”標準”であることを覚えておいてください)。原子3に結合しているメチル基の動きは、大きく動いている最初のフレームから小さく動いた2番目のフレーム、全く動いていない3番目のフレームまで多岐にわたっています。
2段目では、原子2を固定する一方原子1および3に他の選択肢を適用したものです。これらの図を比較するだけでなく実験することで、各選択肢による相違を明快にしかつ精通するようになるでしょう。
原子価の付加と削除
Add Valenceボタンを選択すると、マウスでクリックした原子に追加の原子価(水素原子として表示される)が取り付けられます。新しい原子は、クリックした原子に結合している他の原子からなるべく離れたところに置かれます。
同様に、Delete Atomボタンは原子と開いた原子価を消去します。これを用いるとき、選択した原子とそれに結合している全ての単原子価(水素原子)が取り除かれます。結合上をクリックすることで、ダングリングボンドの消去にも用いられます(図19)。
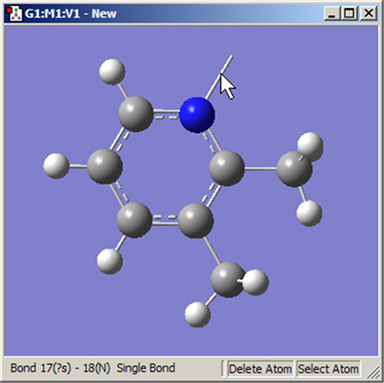
図19.ダングリングボンド(開いた原子価)の消去
Delete Atomボタンは、原子とダングリングボンドを消去するのに用いられます。後者の例を分子の上部に示します。クリックすると開いた原子価を消去します。
原子に対しての反転(鏡面反転)
Invert About Atomボタンにより、選択した原子に対して分子の対称性を反対にさせることが出来ます。図20はこの機能を用いたところを示しています。
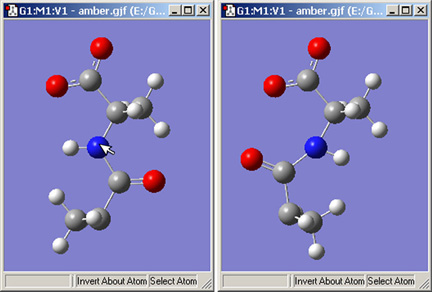
図20.原子に対する構造の反転
Invert About Atomツールを選択して窒素原子をクリックすると、右の構造が得られます。
構造のクリーニング
CleanボタンおよびEdit=>Cleanメニュー項目は共に、決められたルールの組に基づいて、化学的直感により合致するように分子の構造を調整します。得られた結果はあくまでも近似であり、完璧だという意味ではありません。構造は、遷移状態のような非古典的な場合には、調整を必要とします。
GaussViewのクリーン機能は、Clean Controls環境設定パネル(図21)によってカスタマイズできます。
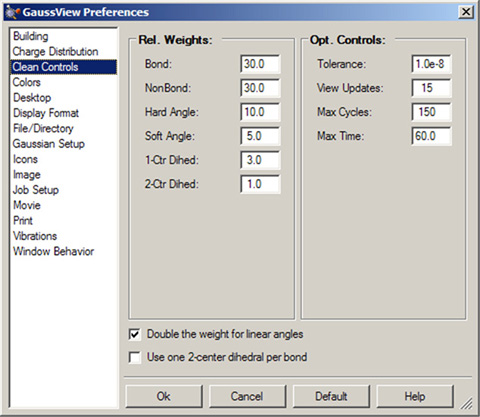
図21.クリーン機能のカスタマイズ
この環境設定パネルで、GaussViewのクリーン機能の動作をカスタマイズすることが出来ます。
デフォルトのクリーン設定は、期待する“標準的な”結果が得られるようにバランスが取られています。Clean Controls環境設定の設定値の変更は、予想外でかつ望ましくない結果を引き起こす可能性がありますので、変更する前に以下のセクションを熟読してください。
以下の制御はダイアログで利用可能です。Rel. Weights 列の各欄は:
- Bond:目標とする結合長は、結合を作っている2つの原子のvan der Waals半径を元に、結合タイプにより微調整を加えることで決定されます。これらは化学的な正確さを意図するものではありませんが、Gaussianで実行する上で妥当な入力を与えます。
- NonBond:直接結合していない全ての原子間の反発項。この要素を導入する目的は、原子中心が近付かないようにすることで、ある原子が他の原子を隠さないようにすることです。しかしながら、ビフェニルについては調節しません。
- Hard Angle:Hard angleは2-4つ結合を有する原子中心に対して計算されます。目標とする角度がわかっているので、より大きな重み付けをします。これらの項は、原子中心の整合性を保持するために極めて重要です。
- Soft Angle:Soft angleは5つ以上の数の結合を有する原子中心に対して計算されます。目標とする角度はわかっていなくても、規範に従えば、望ましい結果に達することが出来ます。三角両錘構造を作るためには、アキシャルの結合角を180度に調節します。直線状結合角にバイアスをかけるオプションがオンになっていることを確認してください。ほとんどの場合、クリーンの手続きは望ましい構造を生成するでしょう。より複雑な配位構造をクリーニングするこつをつかむには、いろいろな配位のパターンで実践してみてください。
- 1-Ctr Dihed:ニューマン投影から計算し、結合を減らして行って5つ未満になるようにしたもの。この項は、2-center dihedralsが歪みを生じるとき、原子中心の整合性を保持するのに役立ちます。
- 2-Ctr Dihed:これは3級構造にのみ影響を及ぼす項です。目標となる2面角は、結合のタイプにより重なり型かねじれ型になります。各原子が多重結合を含んでいれば、目標となる2面角は重なり型です。
ウィンドウ下部にあるチェックボックにスは、以下の効果があります:
- Double the weight for linear angles:この項は、原子中心の角度がよくわからないsoft angleのクリーンの助けとなります。
- Use one 2-center dihedral per bond:通常、このオプションはチェックされていません。計算の遅いシステム上で、より良い実行性能を提供するのに有用です。
Opt. Controls列の各欄は:
- Tolerance:より低い値を指定するとより正確な構造が得られますが、計算時間が著しく長くなります。指定範囲は1.0E-5から1.0E-12の間です。
- View Updates:クリーンされている分子の表示をどのくらいの間隔で更新するかを制御します。1を指定すると、クリーンのサイクルごとに毎回更新します。10を指定すると、クリーン10サイクルごとに更新します。0を指定すると、クリーンが完了するまで表示を更新しません。システムによっては、この値はクリーンの実行時間に大きな影響を与えます。
- Max Cycles:クリーンでの最適化の最大ステップ数。推奨値は150です。より大きな構造ではより大きな値を指定する必要があるかもしれません。
- Max Time:クリーン操作完了までに許容される最長時間(秒)
クリーンで用いられる力場のさまざまな構成要素はそれぞれ関連しており、それを念頭に置くことが重要です。ある重みを変更すると、他の重みの振舞いに影響を及ぼします。例えば、非結合への過大な重み付けは長い結合を生じます。同様に、Hard Angleへの過大な重み付けは2-center dihedralsに影響を及ぼします。
重みを0.0に設定することで、各項を無効にすることが出来ます。例として、1-center dihedral項を無効にするとクリーンがより早く終わりますが、得られた3級構造は良くないでしょう。bondおよびhard angle項の重みは無効にしないでください。前者は、他の項に比べて高い状態に保たなければなりません。
クリーンの性能向上のためのヒント
クリーン機能の実行が使用されているシステム上で極端に遅い場合は、以下の設定を試してください:
- NonBond: 0
- 1-Ctr Dihed: 0
- Use one 2-center dihedral per bond:チェック
- View Updates: 0
クリーン機能の実行性能の貧弱さは、システム上のメモリ不足が原因かもしれません。
結合の再構成
RebondボタンもしくはEdit=>Rebondメニューにより、結合した原子同士をGaussViewで再認識するための、距離アルゴリズムに基づいた結合の再構成プロセスを開始します。Gaussianでは、画面に表示されている結合情報は計算には使用していないことに留意してください。この情報は、分子の化学的性質を見やすくするために提供されています。
対称性の指定
Point Group Symmetryダイアログは分子構造に対して望ましい対称性を指定するのに用いられます(図22)。Edit=>Point Groupメニューにより表示されます。それぞれの項目には以下の意味があります:
- Enable point group symmetry:GaussViewの対称操作の機能を使用可能にする。
- Constrain to subgroup:構造を拘束するための点群を選択
- For:All changesを選択すると以降の構造操作に対して対称性を課す
- Approximate higher-order point groups:適用し得るより高い対称性の部分点群
- Tolerance:構造パラメータを等しいと見なすカットオフレベル
- Symmetrize button:分子構造に対して選択したより高次の点群を速やかに課す
- Always track point group symmetry:GaussViewで、構造が変化するごとに連続的に点群同定の計算を行うよう指定する。構造パラメータを調整する(例えば、結合長スライダーを使う)ごとに点群が連続的に変化することを観察するためには、対称性拘束の設定のNoneと併せてこの項目を使用します。通常、点群は構造変化が完了した後だけ特定されます。動作の遅いシステムで非常に大きな分子を扱う場合、この項目を有効にすることで応答時間の顕著な遅れを引き起こすかもしれません。
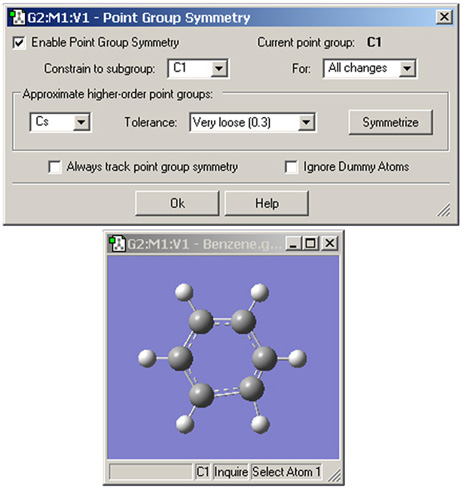
図22.分子構造への点群の指定
この例では、GaussViewはこの歪んだベンゼン分子の現在の点群をC1と特定しています。この構造からは、C2v点群も適切であることがわかります。Symmetrizeボタンをクリックすることで、指定した点群が構造に対して適用されます。Tolerance欄は対称性の特定プロセスをきつくするまたは緩めることに用いることが出来ます。この場合、許容範囲を緩めることでGaussViewは最も高い点群であるD6hの特定が可能になります。
Symmetrizeボタン(ツールバー上)およびEdit=>Symmetrizeメニュー項目を使用して、この分子に対するPoint Group Symmetryダイアログでの最近のTolerance設定(あるいはデフォルトの Normal)に従って、構造に対して特定可能な最大の点群を速やかに課します。分子に対して点群対称性を有効にすることで副作用をもたらす場合もあります(もしあれば、以前の設定を再び用います)。
図23で示されているように、ビューウィンドウのステータスバーには分子に対して特定した対称性および制限を課した対称性が表示されます。
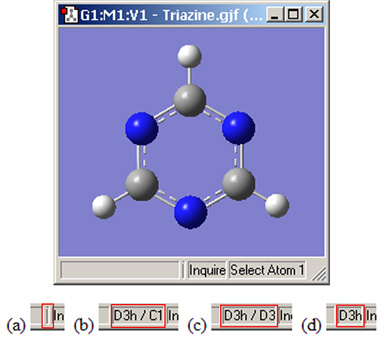
図23.対称性に関するステータスバーの表示
トリアジン分子に対するステータスバー内にあ4つのる対称性情報欄の図:(a)対称性が認識されていない;(b)分子の対称性は認識されている(D3h) が拘束はない(C1);(c)分子の対称性(D3h)および構造をD3対称に拘束;(d)分子の対称性および構造の拘束ともにD3h。
|
