ダイアログ
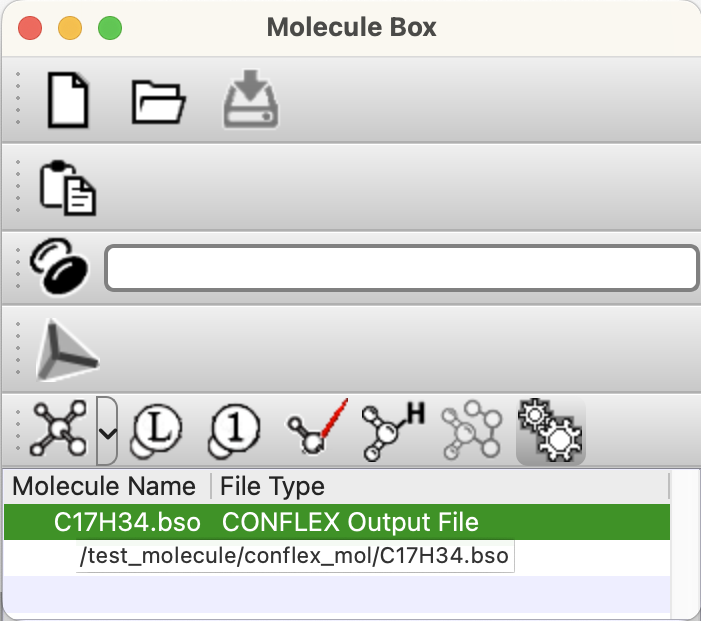
Molecule Boxダイアログ
開いているファイルのリストが表示されます。
表示されている分子ファイル名にマウスカーソルを合わせ数秒待つと、ファイルのフルパスが表示されます。
ツールバーアイコンは、それぞれメニューの項目に対応しています(後述)。
二段目に表示されているプログレスバーは、大きなファイルの読み込みや分子軌道の作成など、長い処理の際に使用されます。
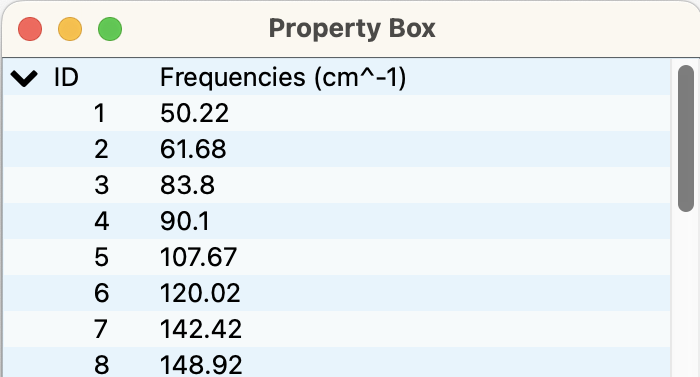
Property Boxダイアログ
開いているファイルのプロパティが表示されます。
開いたファイル種類によって、表示は変わります。
図は、振動の表示例です。結晶探索結果のファイルの場合、ヘッダーをクリックすることにより、ソート可能です。
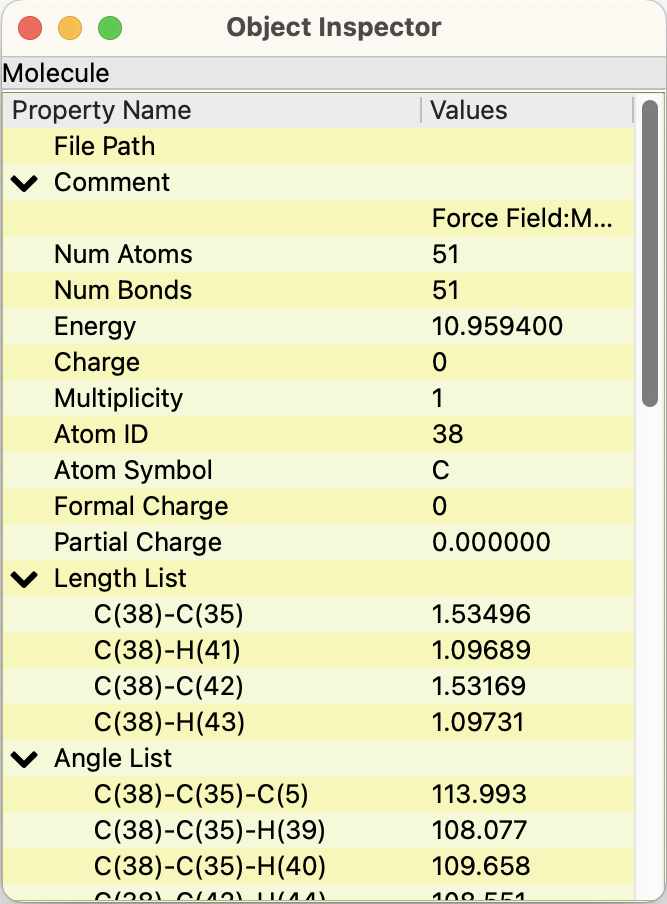
Object inspectorダイアログ
開かれている分子ファイルの情報が表示されています。分子ウィンドウの選択状態に従って、ファイルや原子・結晶の情報を表示します。
図は、原子をクリックした時のものです。
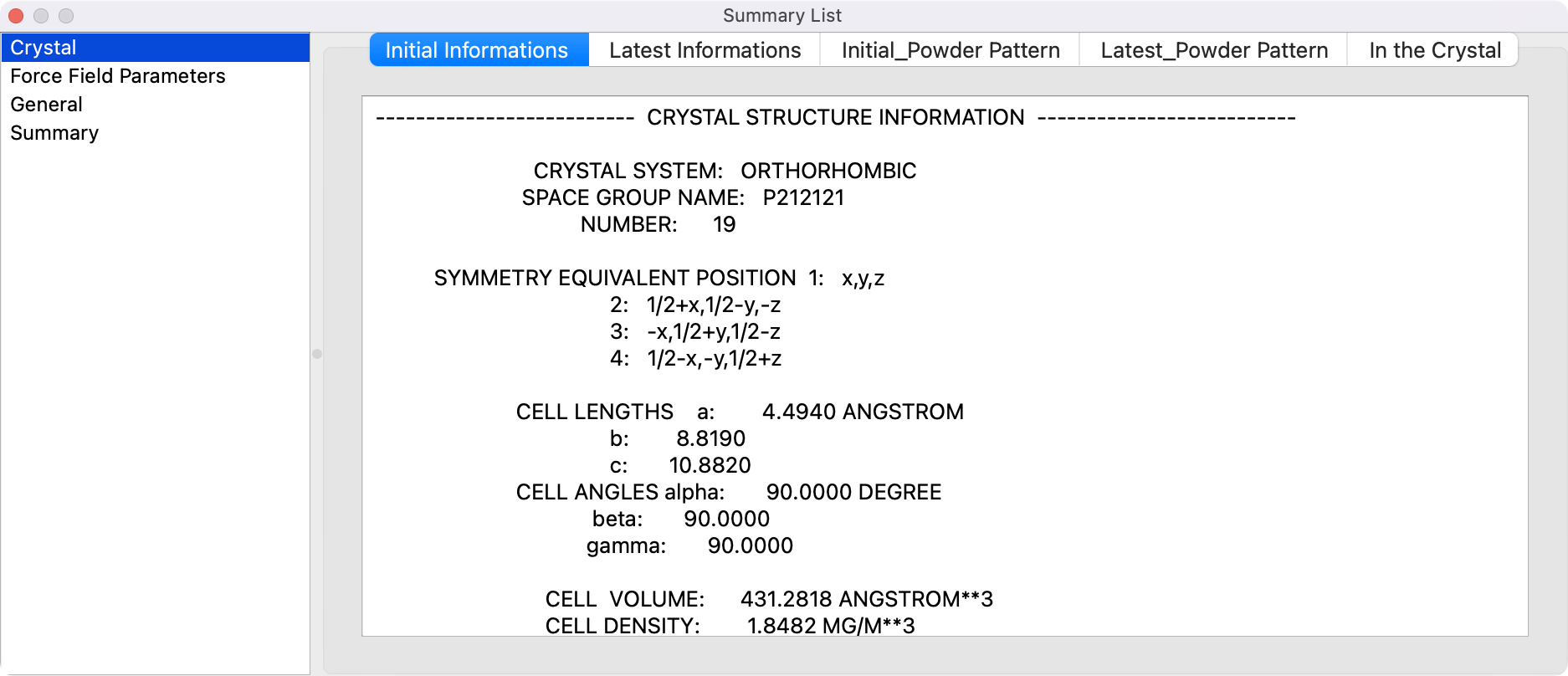
Summary Listダイアログ
計算結果のファイルを開いた際に、計算概要などを表示するダイアログです。
例えば、CONFLEX配座解析後の場合は、配座リストを一覧表示します。
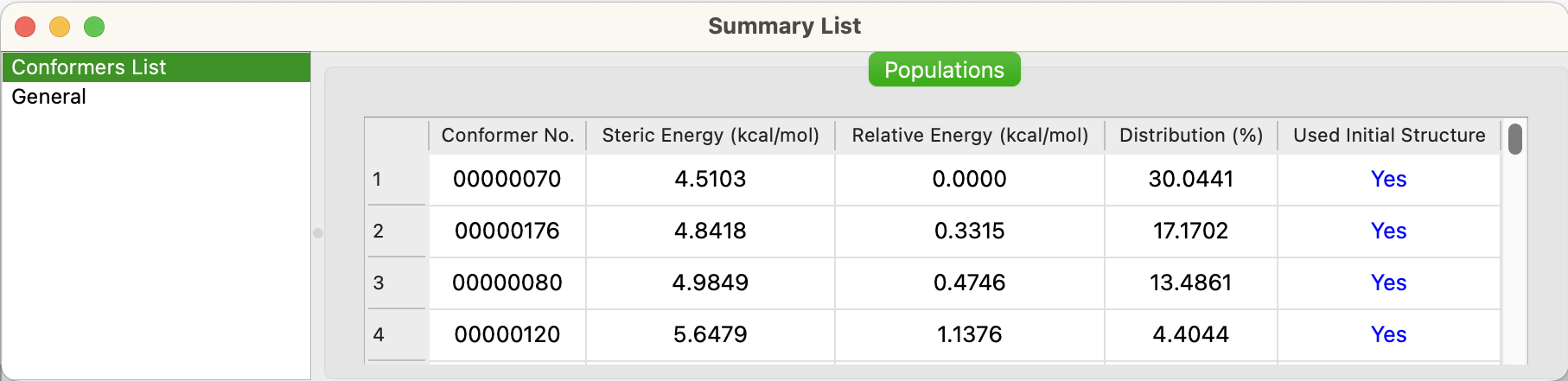
図の配座リストは、エネルギーの低い順に並んでいます。
- Conformer No.
- 配座探索中に、見いだされた順番です。
- Steric Energy
- 分子力場による立体エネルギー。
- Relative Energy
- 最安定配座のエネルギーを0.0とした時の、相対エネルギー。
- Distribution
- 配座立体エネルギーからのボルツマン分布での、存在確率。
- Used Initial Structure
- CONFLEX配座解析の出発構造として使用したかどうかの表示。「Search Limit」の範囲内が全て「Yes」となったら、探索終了。
結晶構造最適化後のbsoファイルを開くと、結晶に関する情報が表示されます。
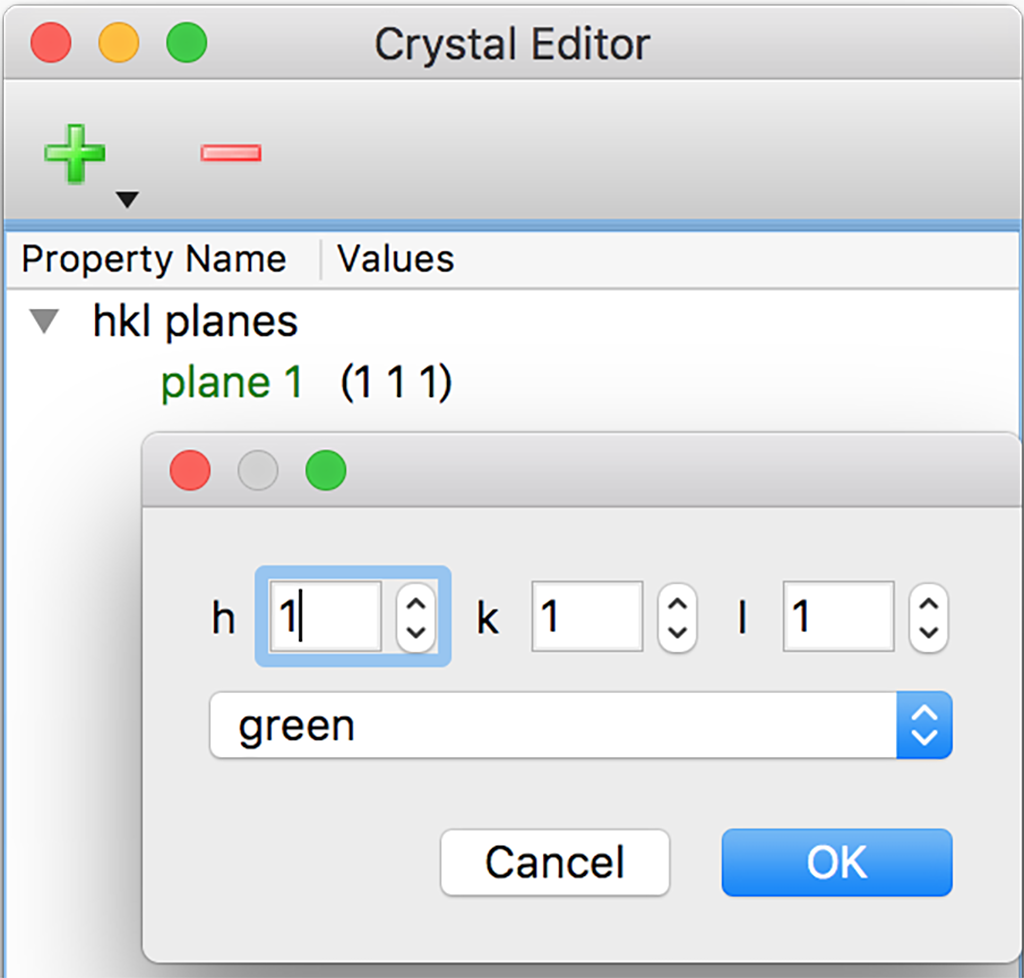
Crystal Editorダイアログ
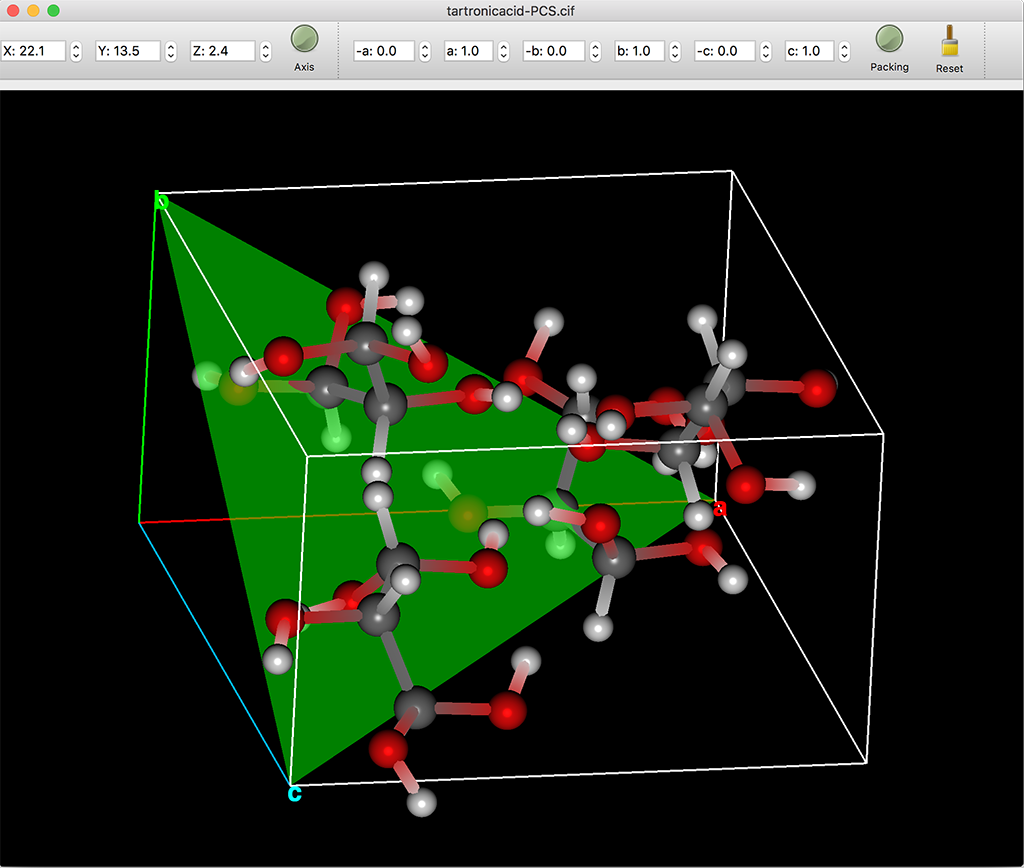
結晶構造表示画面に、結晶面をミラー指数h, k, lで指定して表示させることができます(右図)。 「+」アイコンをクリックすると、ミラー指数ダイアログが開き、各指数および面の表示色を指定して追加できます。 「ー」アイコンをクリックすると、選択した面を削除できます。
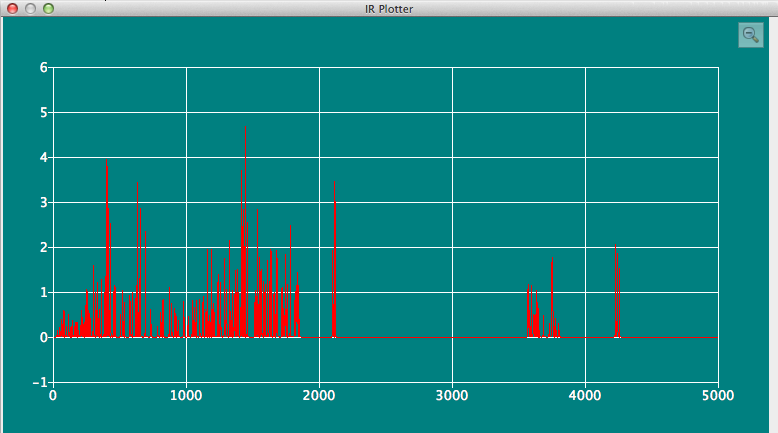
IR Plotterダイアログ
分子ファイル内に振動データが含まれている場合、それをスペクトルとして表示します。 スペクトルの拡大したい部分をマウスでドラッグして囲むと、拡大表示できます。 ダイアログ右上のルーペアイコンをクリックすると、ズームを元に戻したり、再ズームしたりする事も可能です。ズームイン、ズームアウトの比率は、ユーザーがそれまでに行った拡大・縮小の履歴に従って設定されます。
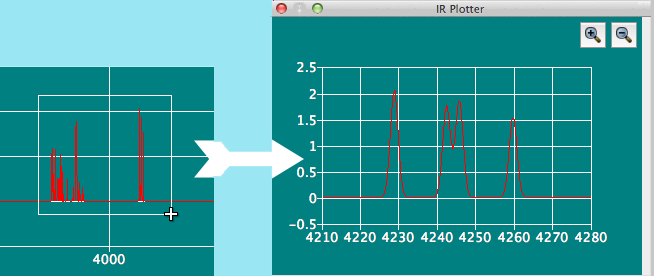
- 対応フォーマット:
- CONFLEX BSO, Gaussian FChkおよびGAMESS/Firefly計算のログファイル
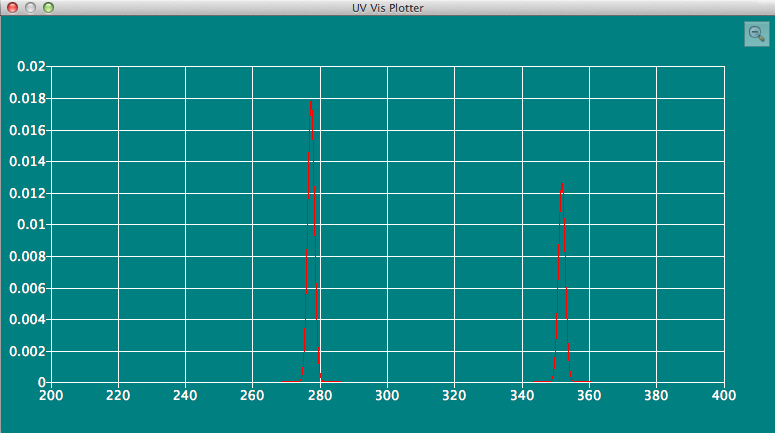
UV/Vis Plotterダイアログ
分子ファイル内に電子励起データが含まれている場合、それをスペクトルとして表示します。
IR Plotterと同様な操作で、ズームイン&アウトが可能です。
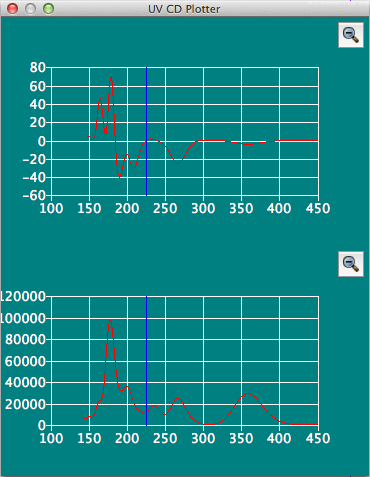
UV CD Plotterダイアログ
分子ファイル(*.bso)内にUV CDデータが含まれている場合、それをスペクトルとして表示します。 CDスペクトルに加えて、UVスペクトルも上下に並べて表示されます。青い縦線は、上下のスペクトルを比較するための基準線として表示しています。この線は、マウスカーソルに追随して移動します。 他と同様に、スペクトルの拡大・縮小操作も可能です。
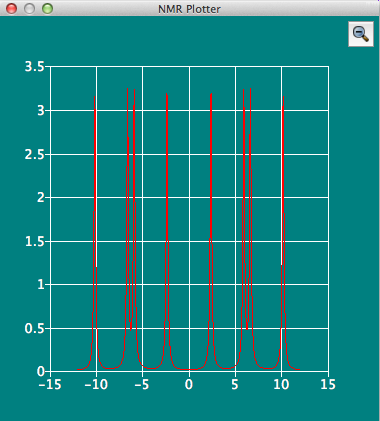
NMR Plotterダイアログ
CONFLEXで行ったNMR 3JHHカップリング定数計算の結果を表示します。分子ファイルが保存されたフォルダー内にNMRデータが含まれているファイルが保存されている場合、自動的に読み込まれます。 プロッターウィンドウにはプロトンNMRのピーク形状が、選択されている水素原子に対して表示されます。目的の水素原子を、分子ウィンドウ内でマウスを用いクリックして選択してください。 他と同様に、スペクトルの拡大・縮小操作が可能です。