AMBER力場による計算
Amber力場による分子の構造最適化について説明します。
【構造最適化と振動解析の実行】
TRP Cageを例として、Amber力場による構造最適化を実行する方法について説明します。
まず、TRP Cageの分子構造データをAmber prmtop形式およびAmber inpcrd形式で、trpcage.prmtop, trpcage.crdとして準備します。
ファイルの作成には、AmberToolsのLEaPプログラムを利用します。詳しい操作方法は、Amberのマニュアルやチュートリアルを参照してください。
TRP Cageのアミノ酸配列は、一文字表記で「NLYIQWLKDGGPSSGRPPPS」です。これを元に、以下の内容のLEaP入力ファイルを作成します(ファイル名は、tleap.inとします):
source leaprc.protein.ff14SB
TRP = sequence { NASN LEU TYR ILE GLN TRP LEU LYS ASP GLY GLY PRO SER SER GLY ARG PRO PRO PRO CSER }
saveAmberParm TRP trpcage.prmtop trpcage.crd
quit
ファイルを作成したら、AMBERの環境が構築されている場所で、以下のコマンドによりtLEaPを実行します。これにより、TRP Cageのトポロジーおよび座標ファイルが作成できます。
tleap -f tleap.inenter
これらtrpcage.prmtop, trpcage.crdファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります (Sample_Files\CONFLEX\amber\trpcage.prmtop.[prmtop|crd])。
ここでは、ファイルフォーマットの詳細は省略します。詳しくはAmberのファイルフォーマットを解説しているページをご覧ください。また、prmtopファイルに関してはこちらのファイルに詳しくの説明されています。
trpcage.prmtopファイルの先頭だけ示します。系内の、原子数・残基数や各原子に割り当てられている原子名などが示されています。このファイルには、シミュレーション中に変化しない情報が含まれています。
%VERSION VERSION_STAMP = V0001.000 DATE = 12/01/20 16:04:37
%FLAG TITLE
%FORMAT(20a4)
NASN
%FLAG POINTERS
%FORMAT(10I8)
304 12 150 160 346 219 700 656 0 0
1701 20 160 219 656 53 124 138 26 0
0 0 0 0 0 0 0 0 24 0
0
%FLAG ATOM_NAME
%FORMAT(20a4)
N H1 H2 H3 CA HA CB HB2 HB3 CG OD1 ND2 HD21HD22C O N H CA HA
CB HB2 HB3 CG HG CD1 HD11HD12HD13CD2 HD21HD22HD23C O N H CA HA CB
HB2 HB3 CG CD1 HD1 CE1 HE1 CZ OH HH CE2 HE2 CD2 HD2 C O N H CA HA
CB HB CG2 HG21HG22HG23CG1 HG12HG13CD1 HD11HD12HD13C O N H CA HA CB
HB2 HB3 CG HG2 HG3 CD OE1 NE2 HE21HE22C O N H CA HA CB HB2 HB3 CG
〜〜〜〜〜〜〜〜〜〜
trpcage.crdファイルの先頭だけ示します。分子に付けられている名前・原子数に続いて、各原子の座標がX, Y, Zの順番に保存されています。
NASN 304 3.3257700 1.5479090 -0.0000016 4.0461540 0.8399910 -0.0000029 2.8230940 1.4995080 -0.8746870 2.8230970 1.4995070 0.8746850 3.9700480 2.8457950 -0.0000001 3.6716630 3.4001290 -0.8898200 3.5769650 3.6538380 1.2321430 2.4969950 3.8010750 1.2413790 3.8774840 3.1157950 2.1311970 4.2537000 5.0171120 1.2321440 5.0052990 5.3404060 0.3150720 3.9848850 5.8179090 2.2659170 4.4080150 6.7337020 2.3147430 3.3596110 5.5042970 2.9944640 5.4855410 2.7052070 -0.0000044 6.0088240 1.5931750 -0.0000084 〜〜〜〜〜〜〜〜〜〜
[Interfaceから実行する場合]
まず、「trpcage.prmtop」ファイルをCONFLEX Interfaceを用いて開きます。ファイルフォーマットは、「AMBER prmtop file」を選択してください。このファイルには座標が含まれていませんので、をクリックすると、もう一度ダイアログが開きます。2番目のダイアログでは、座標が含まれている「trpcage.crd」を選択してをクリックしてください。
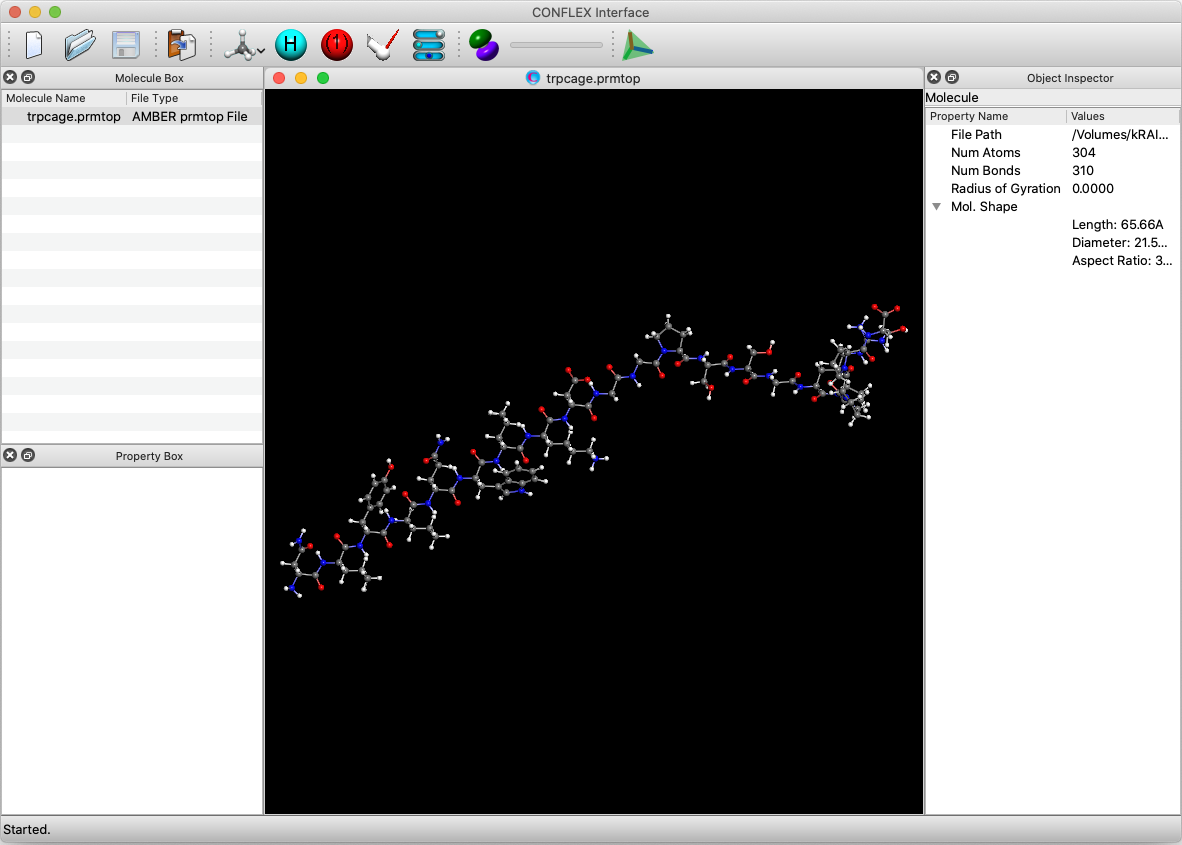
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのボタンをクリックします。デフォルトの構造最適化計算が始まります。分子ファイルがAmberの形式の場合、自動でAMBER力場が選択され計算が開始します。
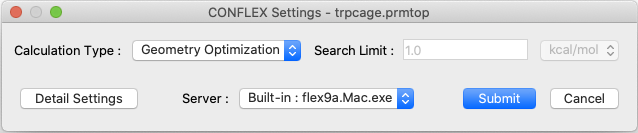
[コマンドラインから実行する場合]
trpcage.prmtop, trpcage.crdをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par trpcageenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【構造最適化計算の出力ファイル】
計算が正常に終了すると、主に以下のファイルが出力されます。
- trpcage.bso
- 計算に用いた力場パラメーターの数値や、各相互作用のエネルギー、更に基準振動解析により得られた熱力学的諸量、振動数、振動モードを出力したファイルです。
- trpcage-F.mol2
- 最適化により得られた構造を、Sybyl-Mol2形式で出力したファイルです。
- trpcage-F.prmtop
- trpcage.prmtopと同じ内容です
- trpcage-F.crd
- 最適化により得られた構造の、AMBER Inpcrd形式の座標ファイルです。
[コマンドラインから実行した場合]
入力ファイルを格納したフォルダに、trpcage.bsoファイルがあります。 これをCONFLEX Interfaceで開くことで、最適化構造を可視化できます。