結晶構造の探索
同一の化学式で表される物質に、複数の結晶構造、結晶多形が存在することがあります。
結晶の物性は結晶構造と密接に関係するため、新規医薬品開発や新規機能性材料開発において、薬の有効性や安全性、また材料の機能性を確保するには、候補化合物の結晶多形スクリーニングが重要になります。
CONFLEXは独自のアルゴリズム[Ishii, H., Obata, S., Niitsu, N. et al. Sci. Rep. 10, 2524 (2020).]により、有機化合物の結晶構造探索と評価を行うことで、結晶多形スクリーニングを実現します。またCONFLEXは、分子のパッキング様式の違いにより生じるパッキング多形や、分子のコンフォメーションの違いにより生じる配座多形のスクリーニングに有効です。
【構造式からの結晶構造探索】
ケンブリッジ結晶学データセンターでは、定期的に、結晶構造予測に関するブラインドテスト (P.M. Lommerse, et al, Acta Cryst. B56, 697-714, 2000)を実施しています。ここでは、ブラインドテストに用いられた5-Cyano-3-hydroxythiophene(II)の結晶構造探索を行う方法について説明します。
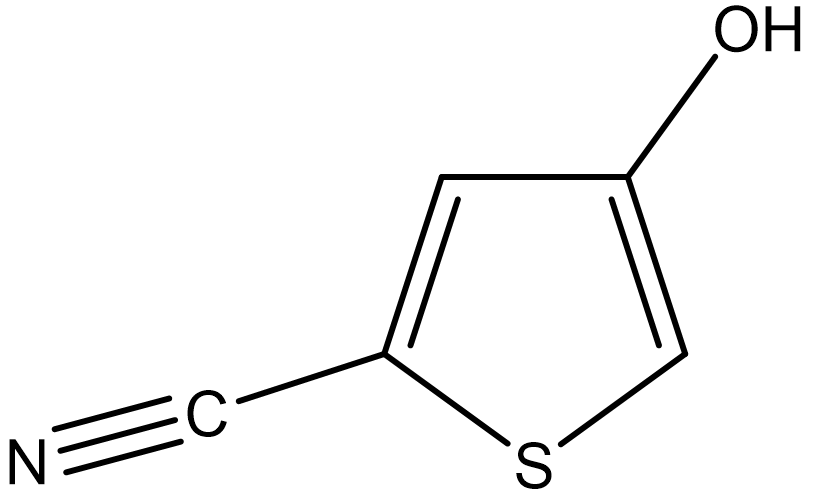
まず、IIの分子構造ファイルを作成します。ここでは、分子構造ファイルの作成に、PerkinElmer社のChemDrawを用います。ChemDrawの利用方法は、マニュアルをご覧ください。
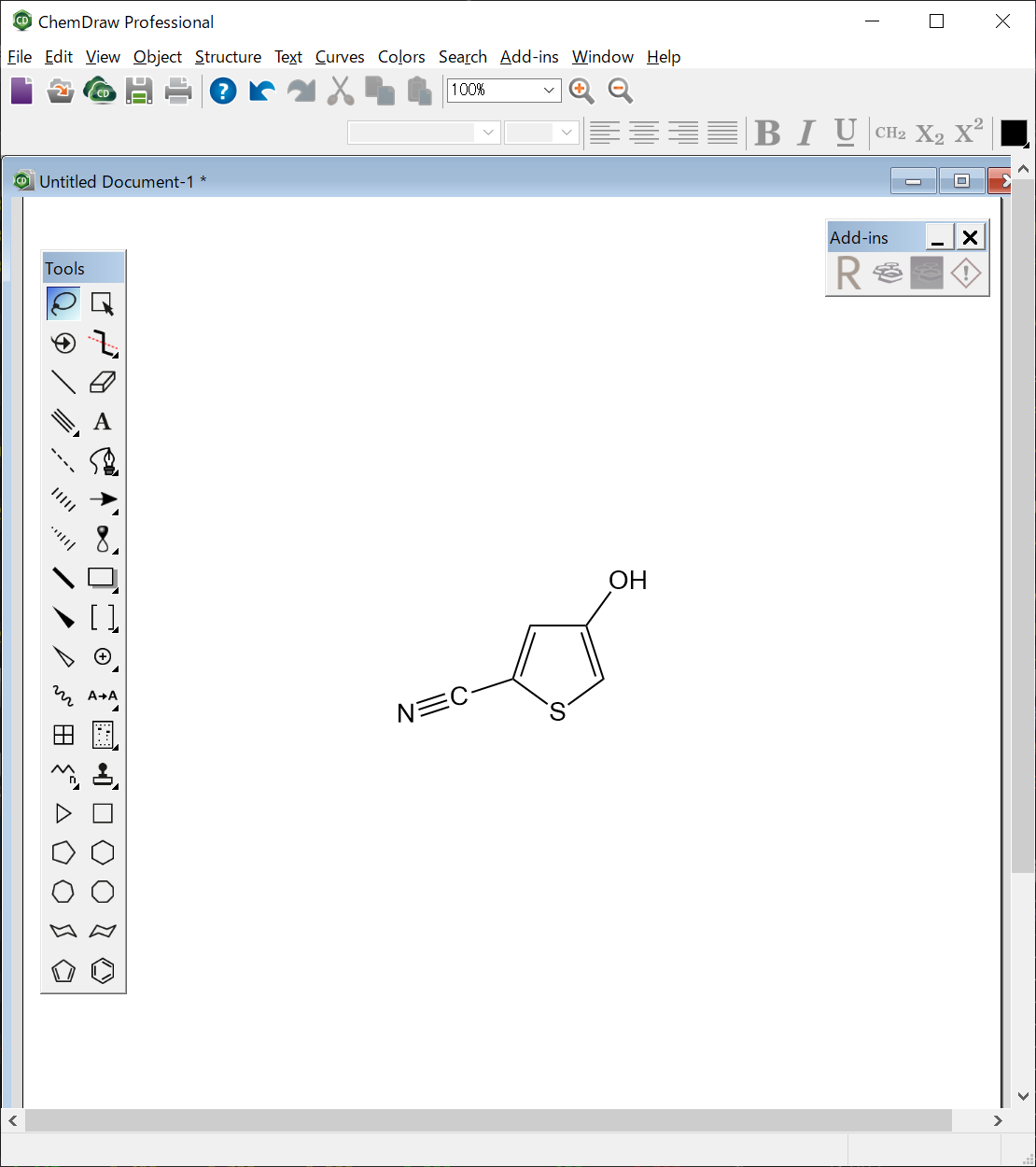
IIの分子構造は、MDL-MOL形式により、“II.mol”として保存します。
II.molファイル
II.mol
ChemDraw08301910352D
8 8 0 0 0 0 0 0 0 0999 V2000
0.2633 0.3011 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0883 0.3011 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0083 -0.4836 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6758 -0.9685 0.0000 S 0 0 0 0 0 0 0 0 0 0 0 0
1.3432 -0.4836 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.5732 0.9685 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.7763 -0.7385 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5732 -0.9520 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 3 2 0
3 4 1 0
4 5 1 0
5 2 2 0
2 6 1 0
3 7 1 0
7 8 3 0
M END
次に、IIの分子構造最適化を行います。
[Interfaceから実行する場合]
II.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。孤立分子の構造最適化が始まります。
[コマンドラインから実行する場合]
II.molをフォルダに格納し、下記コマンドを実行してください。計算が始まります。なお、iniファイルがない場合、CONFLEXは孤立分子の構造最適化をデフォルト設定にて行います。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par IIenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
次に、IIの配座探索を行います。
[Interfaceから実行する場合]
II-F.molファイルをCONFLEX Interfaceを用いて開きます。
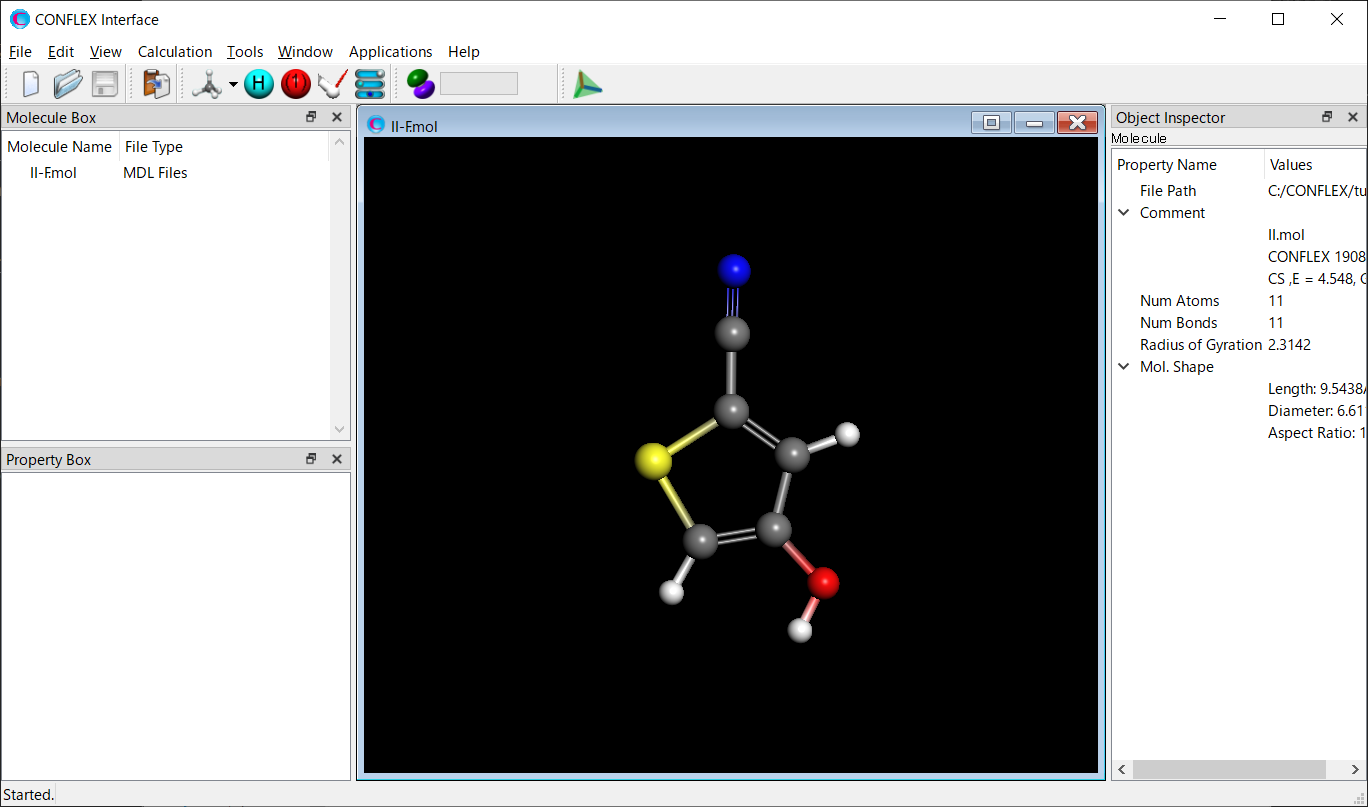
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログの「Calculation Type:」のプルダウンメニューから「Confromation Search」を選択します。「Search Limit:」の値は、「10.0」とします。これは、配座探索空間の範囲を決めます。設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、II-F.iniファイルにキーワードを記述することで行います。
II-F.iniファイル
CONFLEX SEL=10.0
CONFLEXで配座探索を行う場合、「CONFLEXL」キーワードを記述します。
「SEL=10.0」は、配座探索に使用する初期構造のエネルギー範囲(Search Limit)を10.0 kcal/molとすることを意味します。
II-F.molとII-F.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par II-Fenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
配座探索の結果、2つの配座異性体が得られます。ここでは、2番目にエネルギーの低い配座異性体を結晶構造探索計算に用います。そこで、“II-F.sdf”ファイルから、2番目にエネルギーの低い配座異性体の分子構造ファイルを“II-c2.mol”として作成します。II-F.sdfファイルは、II-F.molファイル等と同じフォルダに格納されています。また、Interfaceを利用して配座探索を行った場合、ファイル名が、II-F_conv.sdf、となっています。
II-c2.molファイル
II.mol
CONFLEX 19083010373D 1 1.00000 4.54816 1
CS ,E = 4.548, G = 0.974E-10, P = 48.3970, M( 0), IFN =00000002-00000001
11 11 0 0 999 V2000
1.1214 -0.0104 0.0000 C 0 0 0 0 0
0.7822 -1.3858 0.0000 C 0 0 0 0 0
-0.0000 0.7946 -0.0000 C 0 0 0 0 0
-1.4433 -0.1296 -0.0000 S 0 0 0 0 0
-0.5732 -1.6069 0.0000 C 0 0 0 0 0
1.6924 -2.3749 0.0000 O 0 0 0 0 0
0.0137 2.2245 -0.0000 C 0 0 0 0 0
0.0454 3.3854 -0.0000 N 0 0 0 0 0
2.1374 0.3665 0.0000 H 0 0 0 0 0
-1.1076 -2.5464 0.0000 H 0 0 0 0 0
1.2623 -3.2477 0.0000 H 0 0 0 0 0
1 2 1 0 0
1 3 2 0 0
3 4 1 0 0
4 5 1 0 0
5 2 2 0 0
2 6 1 0 0
3 7 1 0 0
7 8 3 0 0
1 9 1 0 0
5 10 1 0 0
6 11 1 0 0
M END
最後に、結晶構造探索計算を行います。
[Interfaceから実行する場合]
II-c2.molファイルをCONFLEX Interfaceを用いて開きます。
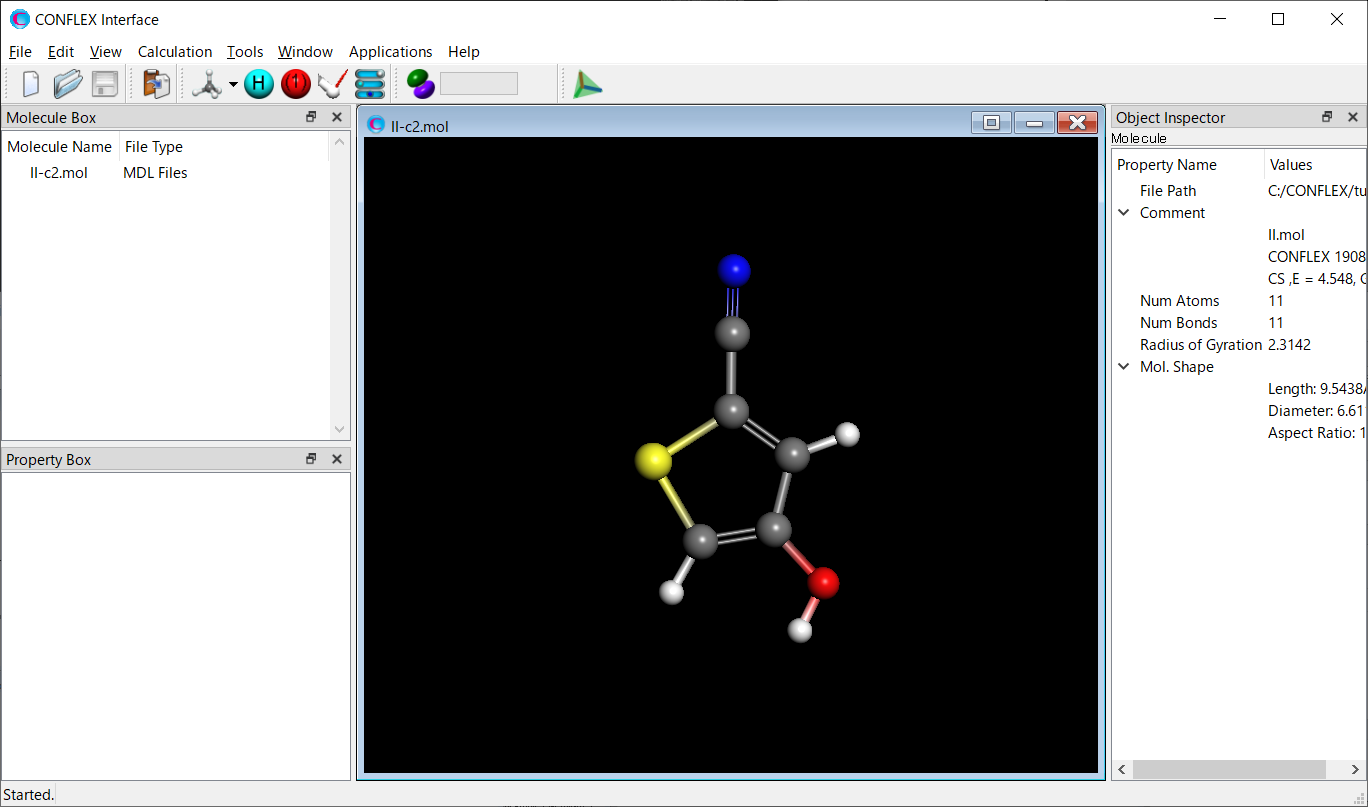
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが開きます。
結晶構造探索計算を実施するために、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから、「Crystal Search」を選択します。
結晶構造の最適化方法は、デフォルトで「ALL」となります。変更は、「Crystal Calculation」ダイアログの「Crystal optimization:」プルダウンメニューからできます。また、本ダイアログでは、カットオフ距離や計算方法等の分子間相互作用計算に関する設定も可能です。
次に、「Crystal Search」ダイアログで、結晶構造探索計算の設定を行います。
まず、探索に利用する空間群を選択するために、「Search Space Group:」のをクリックします。
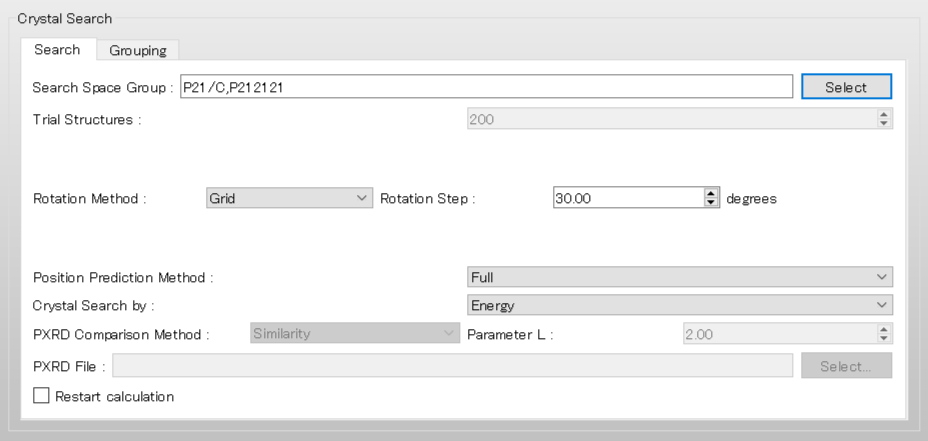
開いたダイアログでは、Cambridge Structural Databaseを用いた統計解析から、有機結晶で多く見られる空間群のTop 10が表示されています。
すべての空間群のチェックボックスにチェックを入れ「OK」をクリックします。
「Rotation Method:」と「Position Prediction Method:」は、分子の回転方法と配置方法を設定します。ここでは、ランダムに決めるとして、両者とも「Random」を選択します。詳細に探索を行いたい場合は、それぞれ「Grid」と「Full」を選択してください。
「Trial Structures:」は、生成する試行結晶構造数を設定します。ここでは、試行結晶構造数は10000とします。試行結晶構造数を多くすることで、探索の信頼性が向上しますが、その分計算負荷は大きくなります。
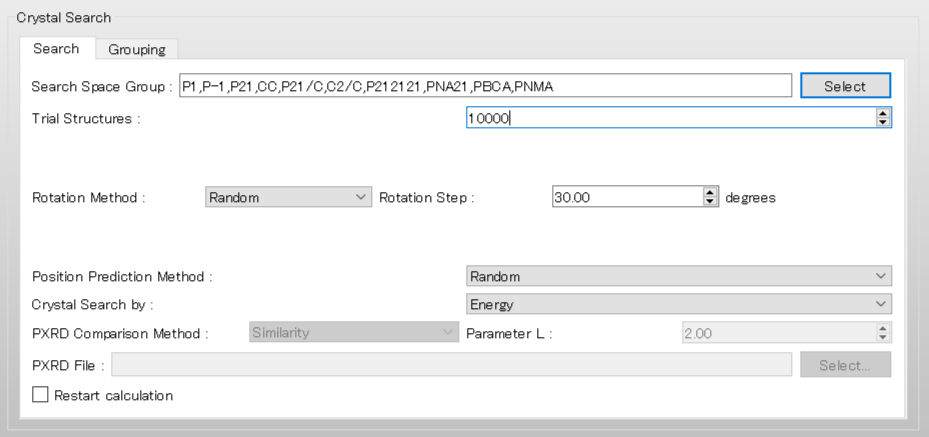
設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、II-c2.iniファイルにキーワードを記述することで行います。
II-c2.iniファイル
CRYSTAL_SEARCH CSP_SPGP=(P21/C,P-1,C2/C,P212121,P21,PBCA,PNA21,PNMA,CC,P1) CSP_ROT_MODE=RANDOM CSP_AUS_MODE=RANDOM CSP_MAX_CRYSTAL=10000 CRYSTAL_OPTIMIZATION=ALL
CONFLEXで結晶構造探索を行う場合、「CRYSTAL_SEARCH」を記述します。
探索に利用する空間群は、「CSP_SPGP=」で設定します。ここでは、P21/c、P-1、C2/c、P212121、P21、Pbca、Pna21、Pnma、Cc、P1を指定します。Cambridge Structural Databaseを用いた統計解析から、有機結晶の約9割が、先に示す空間群に属することが報告されています。
「CSP_ROT_MODE=」と「CSP_AUS_MODE=」は、それぞれ、分子の回転方法と配置方法を設定します。ここでは、それぞれランダムに決めるとして、両者とも「RANDOM」を設定します。詳細に探索を行いたい場合、それぞれ「Grid」と「Full」を設定します。
「CSP_MAX_CRYSTAL=」は、生成する試行結晶構造数を設定します。ここでは、試行結晶構造数は10000とします。試行結晶構造数を多くすることで、予測の信頼性が向上しますが、その分計算負荷は大きくなります。
「CRYSTAL_OPTIMIZATION=」は結晶構造最適化方法を指定します。ここでは、「ALL」を指定し、非対称単位の分子構造、分子配向、分子位置、および格子定数を最適化します。
II-c2.molとII-c2.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par II-c2enter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
計算が終了すると、探索計算の詳細情報が出力されたII-c2.cspファイルが得られます。
II-c2.cspファイル内の「*** PREDICTED CRYSTAL STRUCTURES:」部には、結晶構造探索計算で見つかったIIの結晶多形構造に関する情報が結晶エネルギー順で出力されます。
各構造のデータはII-c2-PCS.cifファイルに出力されています。
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
4 318 1 -15.7921 4.6942 -20.4864 607.6657 1.3663 9.5996 8.3419 10.7545 90.0000 44.8779 90.0000 P21/C 365 4015 20.00 0
30 5295 2 -15.6634 4.7074 -20.3708 306.9963 1.3522 13.4774 7.0951 4.3469 90.0000 132.3915 90.0000 P21 371 4081 20.00 0
147 3622 3 -15.6125 4.7219 -20.3344 614.2783 1.3515 4.3718 19.8181 7.0899 90.0000 90.0000 90.0000 P212121 373 4103 20.00 0
167 3853 4 -15.6061 4.6925 -20.2986 612.8432 1.3547 9.6310 8.3434 7.6267 90.0000 90.0000 90.0000 P212121 325 3575 20.00 0
178 51 5 -15.5986 4.7162 -20.3147 614.9313 1.3501 4.3234 7.1069 21.9413 90.0000 114.1992 90.0000 P21/C 371 4081 20.00 0
190 90 6 -15.4618 4.7170 -20.1789 618.7977 1.3417 4.3746 7.0720 24.0800 90.0000 123.8359 90.0000 P21/C 374 4114 20.00 0
195 1948 7 -15.3029 4.7145 -20.0173 1222.4436 1.3583 14.5999 8.3633 10.4384 90.0000 73.5604 90.0000 C2/C 357 3927 20.00 0
206 169 8 -15.2948 4.7210 -20.0158 612.9536 1.3545 4.0230 8.3785 19.4984 90.0000 111.1490 90.0000 P21/C 358 3938 20.00 0
229 18 9 -15.2940 4.7263 -20.0203 612.5302 1.3554 8.3759 18.2972 8.8552 90.0000 26.8303 90.0000 P21/C 365 4015 20.00 0
290 4 10 -15.2939 4.7218 -20.0156 612.7425 1.3549 8.3793 18.2092 9.2919 90.0000 25.6065 90.0000 P21/C 357 3927 20.00 0
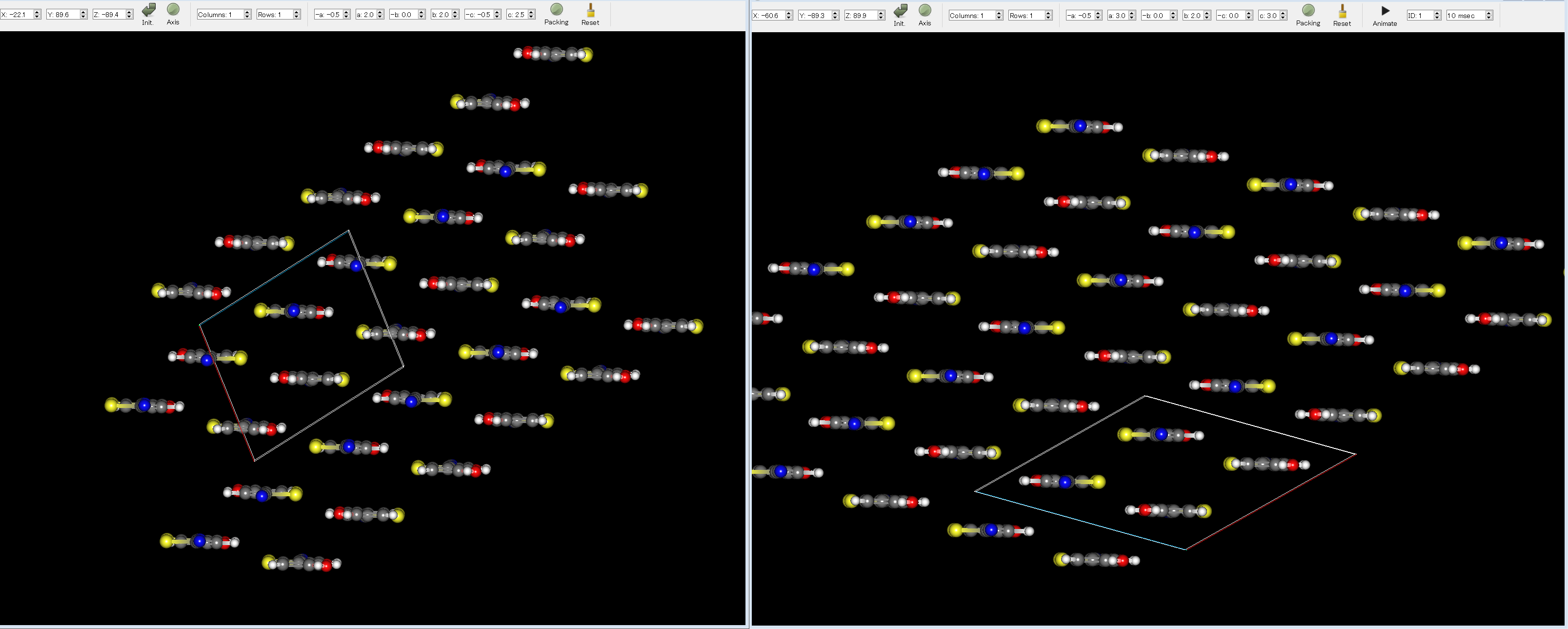
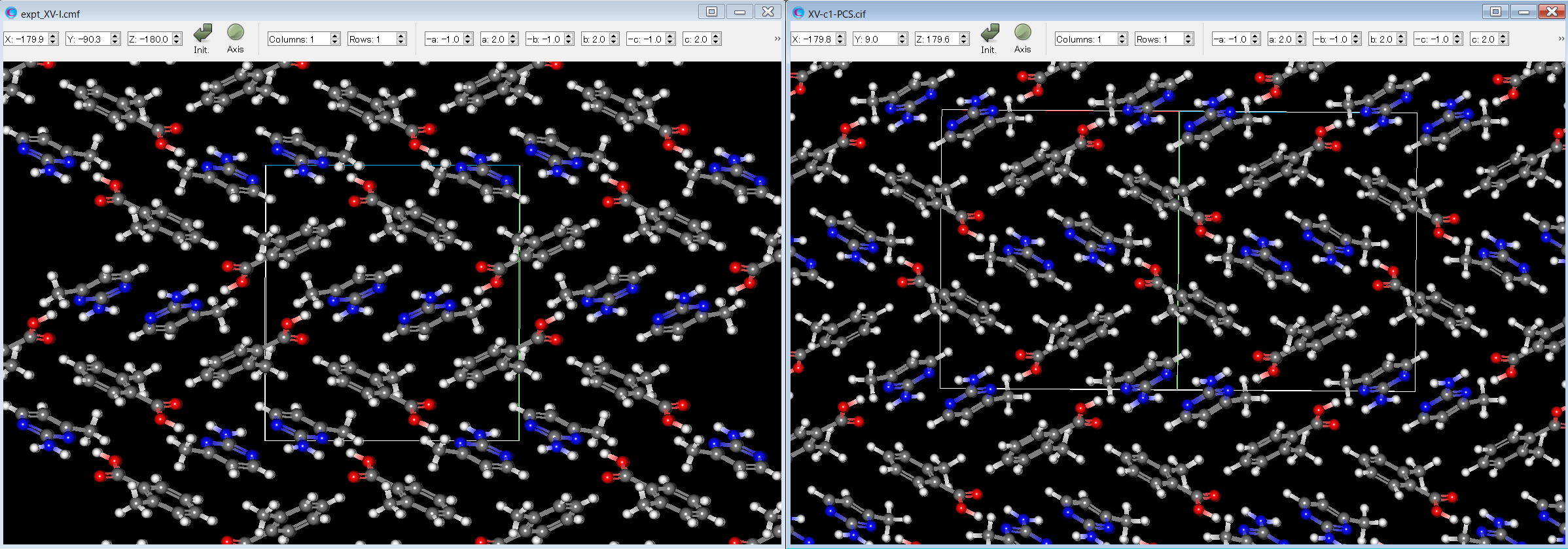
ここで、実験構造(左図)とエネルギー順位1の予測結晶構造(右図)とを比較すると、両者がよく一致することが分かります。
【分子複合体の結晶構造探索】
複数の分子を非対称単位とした場合の結晶構造探索について説明します。
ここでは、ブラインドテストに用いられた2-amino-4-methylpyrimidineと2-methylbenzoic acidの共結晶を取り上げます。
まず、2-amino-4-methylpyrimidineと2-methylbenzoic acidの分子構造ファイルを作成します。分子構造ファイルの作成には、PerkinElmer社のChemDrawを用います。ChemDrawの利用方法は、マニュアルをご覧ください。
2-amino-4-methylpyrimidineの分子構造は、MDL-MOL形式により、“AMP.mol”として保存します。
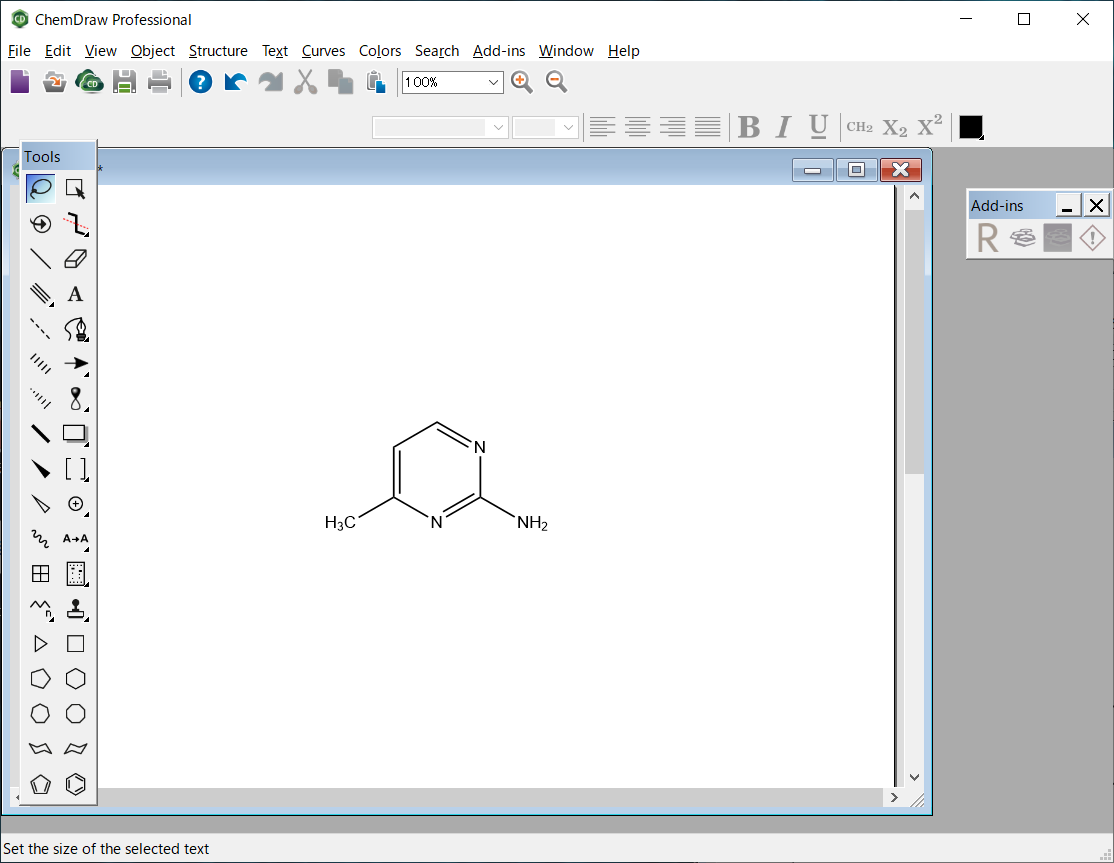
AMP.molファイル
AMP.mol
ChemDraw09022015212D
8 8 0 0 0 0 0 0 0 0999 V2000
-0.7145 0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.7145 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 -0.8250 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
0.7145 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7145 0.4125 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
-0.0000 0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4289 -0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4289 -0.8250 0.0000 N 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
2 7 1 0
4 8 1 0
M END
2-methylbenzoic acidの分子構造は、MDL-MOL形式により、“MBA.mol”として保存します。
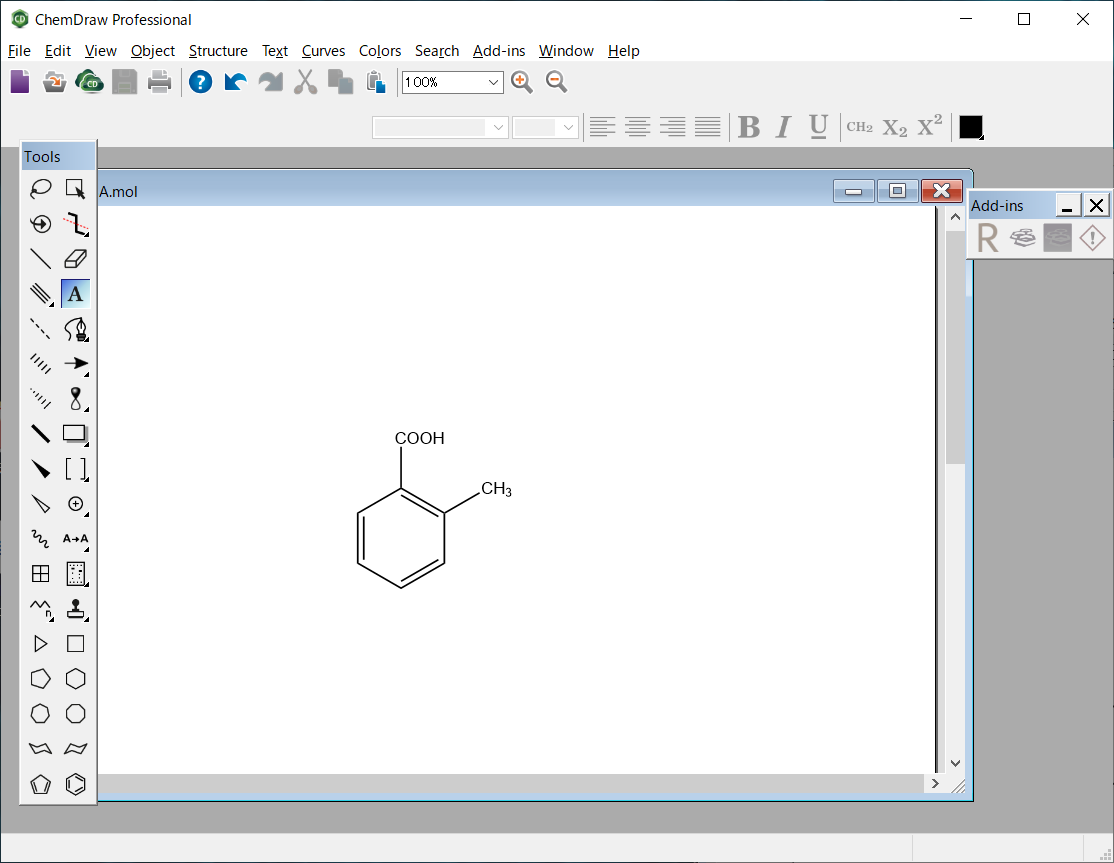
MBA.molファイル
MBA.mol
ChemDraw09022015232D
10 10 0 0 0 0 0 0 0 0999 V2000
-1.0717 -0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.0717 -1.0313 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3572 -1.4438 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3572 -1.0313 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.3572 -0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3572 0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.0717 0.2062 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3572 1.0313 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.0717 1.4438 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
0.3572 1.4438 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
8 9 2 0
8 10 1 0
6 8 1 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
5 7 1 0
M STY 1 1 SUP
M SLB 1 1 1
M SAL 1 3 8 9 10
M SBL 1 1 3
M SMT 1 COOH
M SBV 1 3 0.0000 -0.8250
M END
次に、2-amino-4-methylpyrimidineと2-methylbenzoic acidの分子構造最適化を行います。
[Interfaceから実行する場合]
AMP.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。孤立分子の構造最適化が始まります。
次に、MBA.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。孤立分子の構造最適化が始まります。
[コマンドラインから実行する場合]
AMP.molとMBA.molをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
なお、iniファイルがない場合、CONFLEXは孤立分子の構造最適化をデフォルト設定にて行います。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par AMPenterC:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par MBAenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
2-methylbenzoic acidには、いくつかの配座異性体が考えらえます。そのため、まず、2-methylbenzoic acidの配座探索を行います。
[Interfaceから実行する場合]
MBA-F.molファイルをCONFLEX Interfaceを用いて開きます。
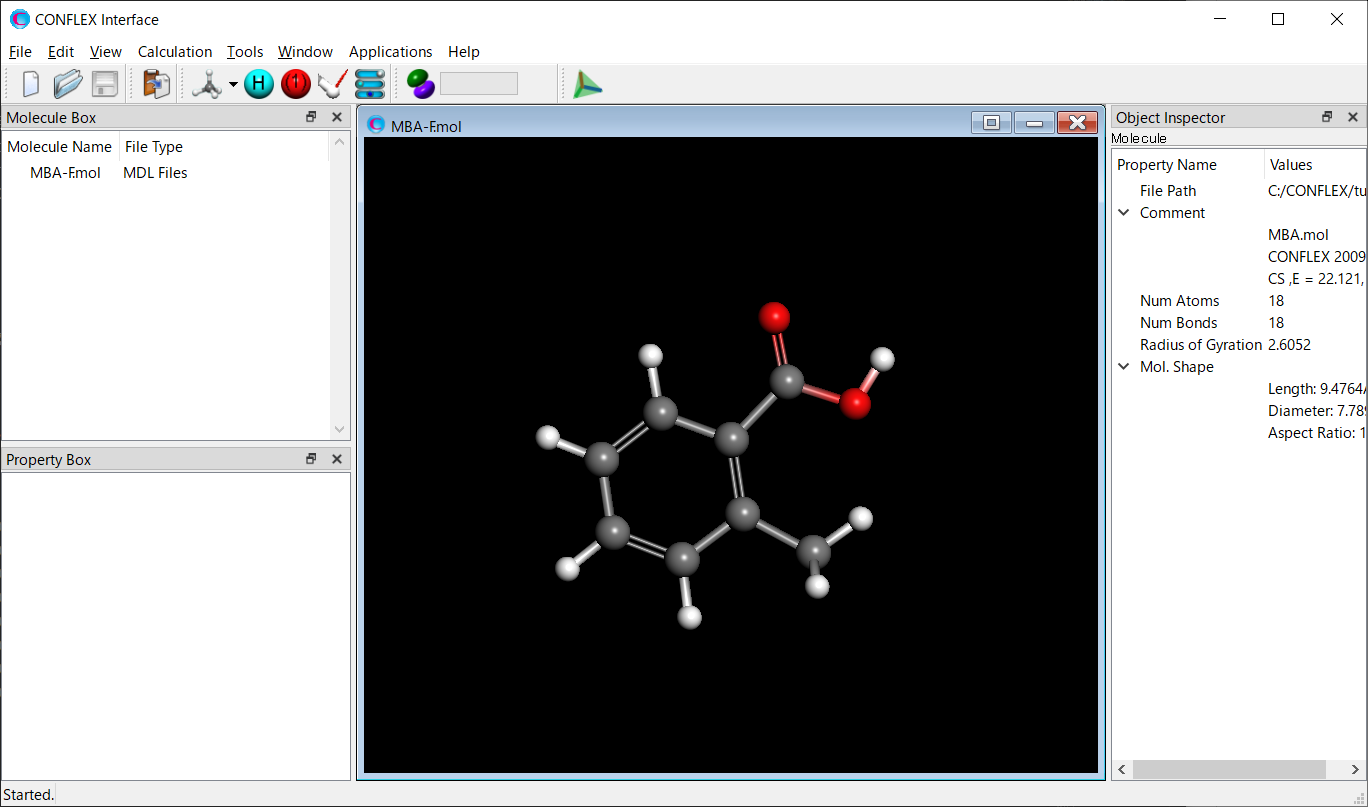
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログの「Calculation Type:」のプルダウンメニューから「Confromation Search」を選択します。「Search Limit:」の値は、「10.0」とします。これは、配座探索空間の範囲を決めます。
設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、MBA-F.iniファイルにキーワードを記述することで行います。
MBA-F.iniファイル
CONFLEX SEL=10.0
CONFLEXで配座探索を行う場合、「CONFLEXL」キーワードを記述します。
「SEL=10.0」は、配座探索に使用する初期構造のエネルギー範囲(Search Limit)を10.0 kcal/molとすることを意味します。
MBA-F.molとMBA-F.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par MBA-Fenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
配座探索の結果、7つの配座異性体が得られます。
ここでは、1番目にエネルギーの低い配座異性体を次の計算に用いることにします。そこで、“MBA-F.sdf”ファイルから、1番目にエネルギーの低い配座異性体の分子構造ファイルを“MBA-c1.mol”として作成します。MBA-F.sdfファイルは、MBA-F.molファイル等と同じフォルダに格納されています。
MBA-c1.molファイル
MBA.mol
CONFLEX 20090215293D 1 1.00000 16.90241 3
CS ,E = 16.902, G = 0.213E-06, P = 89.7913, M( 0), IFN =00000001-00000003
18 18 0 0 999 V2000
-1.3006 0.8083 0.0000 C 0 0 0 0 0
-2.4209 -0.0240 0.0000 C 0 0 0 0 0
-2.2574 -1.4041 0.0000 C 0 0 0 0 0
-0.9754 -1.9534 -0.0000 C 0 0 0 0 0
0.1671 -1.1315 -0.0000 C 0 0 0 0 0
-0.0000 0.2708 0.0000 C 0 0 0 0 0
1.5196 -1.7909 -0.0000 C 0 0 0 0 0
1.1760 1.1942 0.0000 C 0 0 0 0 0
2.3544 0.8930 0.0000 O 0 0 0 0 0
0.8165 2.4930 -0.0000 O 0 0 0 0 0
-1.4568 1.8849 0.0000 H 0 0 0 0 0
-3.4187 0.4075 0.0000 H 0 0 0 0 0
-3.1277 -2.0558 0.0000 H 0 0 0 0 0
-0.8717 -3.0372 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 0.8977 H 0 0 0 0 0
1.4333 -2.8831 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 -0.8977 H 0 0 0 0 0
1.6744 2.9669 -0.0000 H 0 0 0 0 0
8 9 2 0 0
8 10 1 0 0
6 8 1 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
5 7 1 0 0
1 11 1 0 0
2 12 1 0 0
3 13 1 0 0
4 14 1 0 0
7 15 1 0 0
7 16 1 0 0
7 17 1 0 0
10 18 1 0 0
M END
次に、AMP-F.molファイルとMBA-c1.molファイルを用いて、2-amino-4-methylpyrimidineと2-methylbenzoic acid、それぞれ1分子が含まれる分子構造ファイルを“XV.mol”として作成します。XV.molファイルは、AMP-F.molファイルに対して、MBA-c1.molファイルにある原子座標データや結合データを追加して作成しています。XV.molファイルの4行目の「33 33」は、それぞれ、原子数と結合数を示しています。
結合情報「 1 2 2 0 0」は、原子1と原子2が2重結合であることを示します。そのため、2-methylbenzoic acidの結合情報をAMP-F.molファイルに追加する際は、「 8 9 2 0 0」→「 23 24 2 0 0」と原子の通し番号を修正してください。
XV.molファイル
XV.mol
33 33 0 0 999 V2000
1.2096 -1.0822 -0.0000 C 0 0 0 0 0
1.1638 0.2958 0.0000 C 0 0 0 0 0
-0.0000 0.9788 0.0000 N 0 0 0 0 0
-1.1295 0.2628 0.0000 C 0 0 0 0 0
-1.1861 -1.0719 0.0000 N 0 0 0 0 0
-0.0075 -1.7194 -0.0000 C 0 0 0 0 0
2.4311 1.0955 -0.0000 C 0 0 0 0 0
-2.3091 0.9449 -0.0000 N 0 0 0 0 0
2.1397 -1.6334 -0.0000 H 0 0 0 0 0
-0.0748 -2.8030 -0.0000 H 0 0 0 0 0
2.2152 2.1688 0.0000 H 0 0 0 0 0
3.0226 0.8673 0.8921 H 0 0 0 0 0
3.0226 0.8673 -0.8921 H 0 0 0 0 0
-3.1598 0.4121 -0.0000 H 0 0 0 0 0
-2.2706 1.9481 -0.0000 H 0 0 0 0 0
-1.3006 0.8083 0.0000 C 0 0 0 0 0
-2.4209 -0.0240 0.0000 C 0 0 0 0 0
-2.2574 -1.4041 0.0000 C 0 0 0 0 0
-0.9754 -1.9534 -0.0000 C 0 0 0 0 0
0.1671 -1.1315 -0.0000 C 0 0 0 0 0
-0.0000 0.2708 0.0000 C 0 0 0 0 0
1.5196 -1.7909 -0.0000 C 0 0 0 0 0
1.1760 1.1942 0.0000 C 0 0 0 0 0
2.3544 0.8930 0.0000 O 0 0 0 0 0
0.8165 2.4930 -0.0000 O 0 0 0 0 0
-1.4568 1.8849 0.0000 H 0 0 0 0 0
-3.4187 0.4075 0.0000 H 0 0 0 0 0
-3.1277 -2.0558 0.0000 H 0 0 0 0 0
-0.8717 -3.0372 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 0.8977 H 0 0 0 0 0
1.4333 -2.8831 -0.0000 H 0 0 0 0 0
2.0811 -1.5139 -0.8977 H 0 0 0 0 0
1.6744 2.9669 -0.0000 H 0 0 0 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
2 7 1 0 0
4 8 1 0 0
1 9 1 0 0
6 10 1 0 0
7 11 1 0 0
7 12 1 0 0
7 13 1 0 0
8 14 1 0 0
8 15 1 0 0
23 24 2 0 0
23 25 1 0 0
21 23 1 0 0
16 17 2 0 0
17 18 1 0 0
18 19 2 0 0
19 20 1 0 0
20 21 2 0 0
21 16 1 0 0
20 22 1 0 0
16 26 1 0 0
17 27 1 0 0
18 28 1 0 0
19 29 1 0 0
22 30 1 0 0
22 31 1 0 0
22 32 1 0 0
25 33 1 0 0
M END
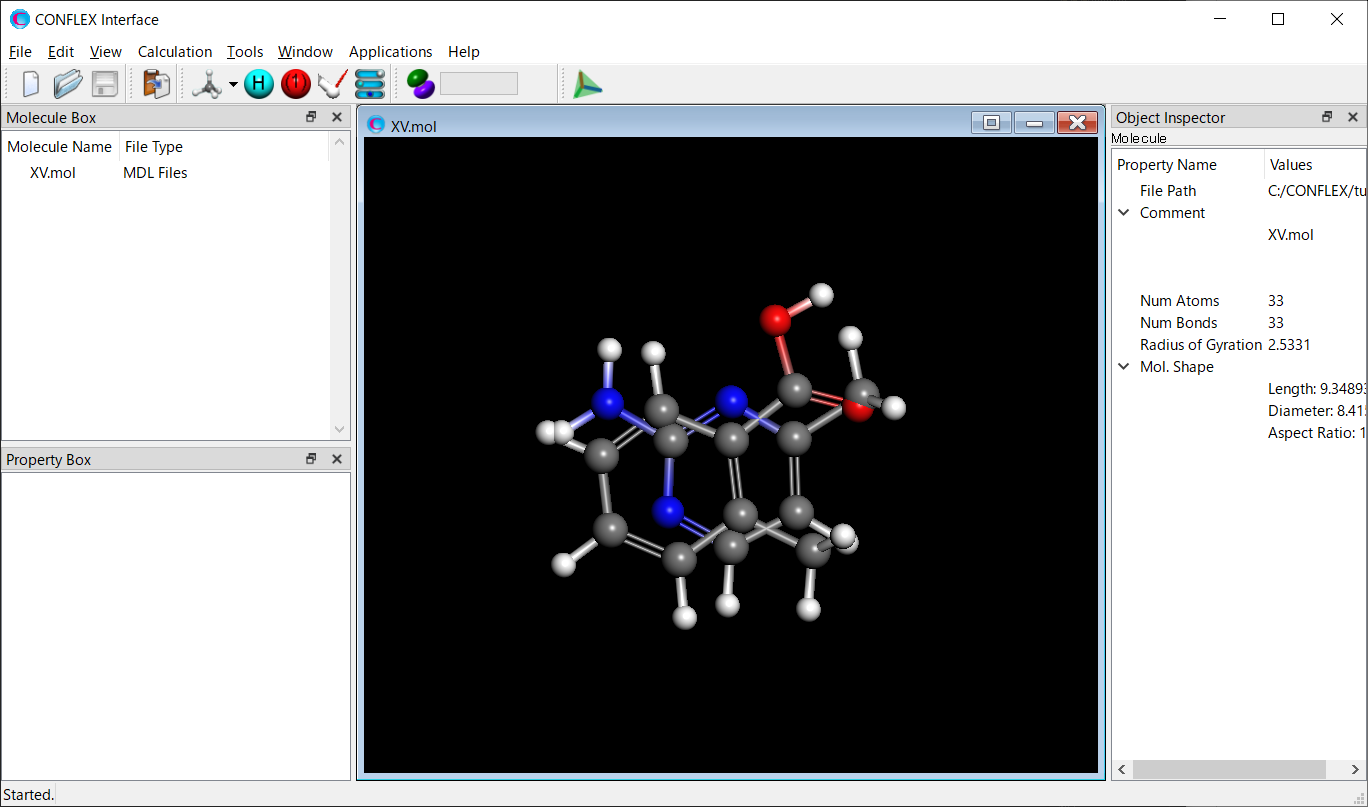
次に、2-amino-4-methylpyrimidine分子と2-methylbenzoic acid分子の初期配置を決めるため、ホスト-リガンド配位探索を行います。
[Interfaceから実行する場合]
XV.molファイルをCONFLEX Interfaceを用いて開きます。この時点で、両分子は重なって存在していますが、配位探索の初期構造生成時に両分子の配置は変更されるので問題ありません。詳しくは「ホスト−リガンド配位探索」を参照してください。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが開きます。
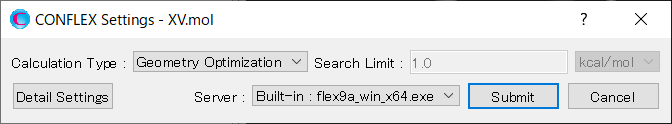
ホスト-リガンド配位探索計算を実施するために、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから、「Host Ligand Search」を選択します。
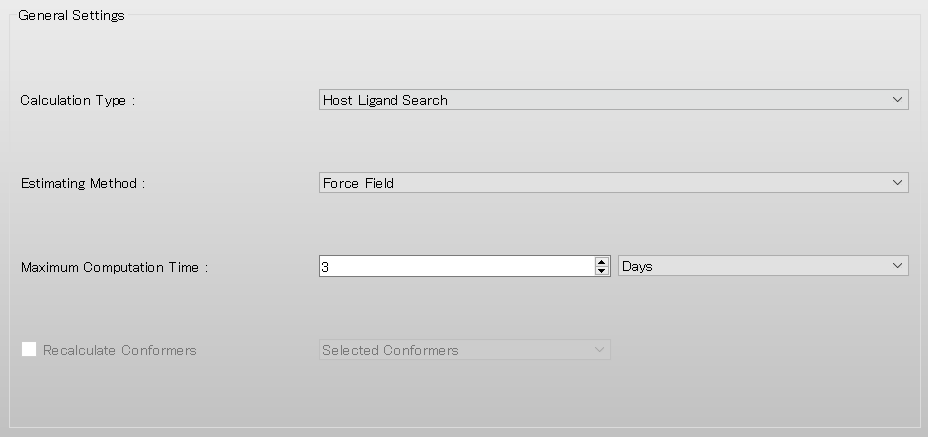
ホスト-リガンド配位探索計算の設定は「Host Ligand Search」ダイアログで行います。
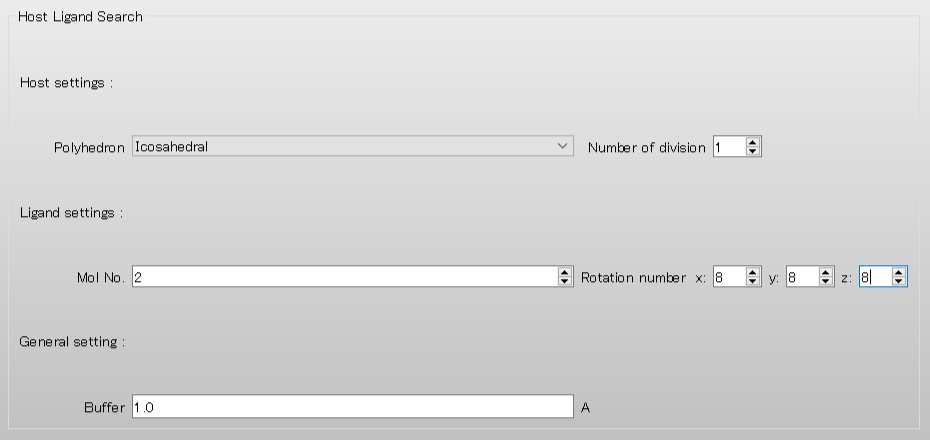
ここでは、リガンドとして設定される2-methylbenzoic acid分子を45度刻みで回転させるため、「Rotational number x, y, z」の値をそれぞれ「8」とします。設定が終わりましたら、をクリックしてください。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、XV.iniファイルにキーワードを記述することで行います。
XV.iniファイル
HLSEARCH HLSEARCH_LIGAND_ROT=(8,8,8)
CONFLEXでホスト-リガンド配位探索を行う場合、「HLSEARCH」キーワードを記述します。
また、リガンドとして設定される2-methylbenzoic acid分子を45度刻みで回転させるため、「HLSEARCH_LIGAND_ROT=(8,8,8)」キーワードを記述します。
XV.molとXV.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par XVenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
ホスト-リガンド配位探索の結果、47の配位構造が得られます。ここでは、1番目にエネルギーの低い配位構造を結晶構造探索計算に用います。そこで、“XV.sdf”ファイルから、1番目にエネルギーの低い配位構造の分子構造ファイルを“XV-c1.mol”として作成します。XV.sdfファイルは、XV.molファイル等と同じフォルダに格納されています。
XV-c1.molファイル
XV.mol
CONFLEX 20090215503D 1 1.00000 -86.91814 17
NONE,E = -86.918, G = 0.249E-06, P = 20.9930, M( 0), IFN =00000001-00000017
33 33 0 0 999 V2000
-4.6389 1.0672 0.0877 C 0 0 0 0 0
-3.2840 1.2059 0.3058 C 0 0 0 0 0
-2.4142 0.1779 0.1589 N 0 0 0 0 0
-2.9370 -1.0122 -0.1834 C 0 0 0 0 0
-4.2342 -1.2388 -0.4067 N 0 0 0 0 0
-5.0617 -0.1892 -0.2687 C 0 0 0 0 0
-2.7235 2.5426 0.6933 C 0 0 0 0 0
-2.0889 -2.0671 -0.3182 N 0 0 0 0 0
-5.3328 1.8902 0.1921 H 0 0 0 0 0
-6.1102 -0.3980 -0.4587 H 0 0 0 0 0
-1.6892 2.4500 1.0374 H 0 0 0 0 0
-2.7441 3.2203 -0.1656 H 0 0 0 0 0
-3.3069 2.9800 1.5099 H 0 0 0 0 0
-2.4812 -2.9552 -0.5738 H 0 0 0 0 0
-1.0952 -1.9477 -0.1563 H 0 0 0 0 0
2.8428 1.4208 -0.4637 C 0 0 0 0 0
4.1800 1.8122 -0.5390 C 0 0 0 0 0
5.1849 0.9050 -0.2233 C 0 0 0 0 0
4.8550 -0.3923 0.1698 C 0 0 0 0 0
3.5126 -0.8046 0.2572 C 0 0 0 0 0
2.4937 0.1165 -0.0682 C 0 0 0 0 0
3.2239 -2.2160 0.6905 C 0 0 0 0 0
1.0518 -0.2696 -0.0173 C 0 0 0 0 0
0.6010 -1.3998 0.0329 O 0 0 0 0 0
0.2426 0.8063 -0.0191 O 0 0 0 0 0
2.0730 2.1442 -0.7236 H 0 0 0 0 0
4.4339 2.8231 -0.8471 H 0 0 0 0 0
6.2281 1.2054 -0.2833 H 0 0 0 0 0
5.6587 -1.0857 0.4113 H 0 0 0 0 0
2.7876 -2.7870 -0.1349 H 0 0 0 0 0
4.1382 -2.7349 0.9990 H 0 0 0 0 0
2.5491 -2.2242 1.5524 H 0 0 0 0 0
-0.6842 0.4546 0.0376 H 0 0 0 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
2 7 1 0 0
4 8 1 0 0
1 9 1 0 0
6 10 1 0 0
7 11 1 0 0
7 12 1 0 0
7 13 1 0 0
8 14 1 0 0
8 15 1 0 0
23 24 2 0 0
23 25 1 0 0
21 23 1 0 0
16 17 2 0 0
17 18 1 0 0
18 19 2 0 0
19 20 1 0 0
20 21 2 0 0
21 16 1 0 0
20 22 1 0 0
16 26 1 0 0
17 27 1 0 0
18 28 1 0 0
19 29 1 0 0
22 30 1 0 0
22 31 1 0 0
22 32 1 0 0
25 33 1 0 0
M END
最後に、結晶構造探索計算を行います。
[Interfaceから実行する場合]
XV-c1.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが開きます。
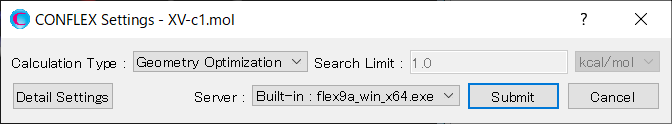
結晶構造探索計算を実施するために、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから、「Crystal Search」を選択します。
結晶構造の最適化方法は、デフォルトで「ALL」となります。変更は、「Crystal Calculation」ダイアログの「Crystal optimization:」プルダウンメニューからできます。
また、本ダイアログでは、カットオフ距離や計算方法等の分子間相互作用計算に関する設定も可能です。
次に、「Crystal Search」ダイアログで、結晶構造探索計算の設定を行います。
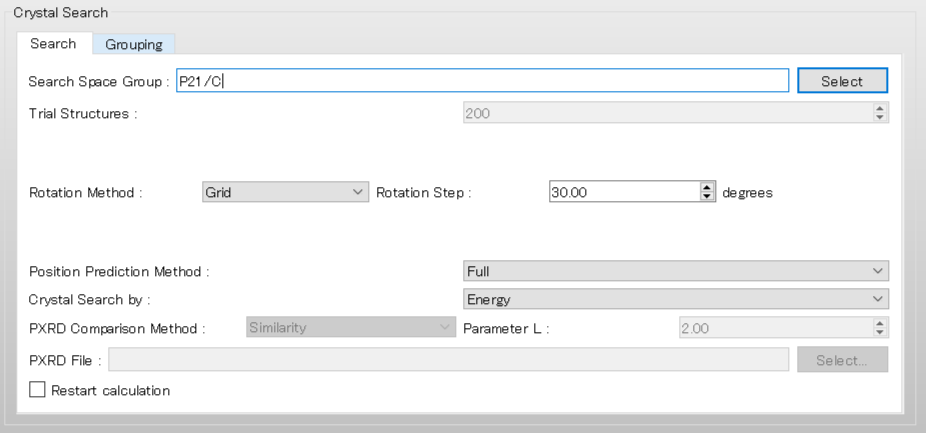
探索に利用する空間群をP21/cとするため、「Search Space Group:」を「P21/C,P212121」から「P21/C」に編集します。また、「Rotation Step:」を20.00とし、分子を回転させる角度の刻み幅を20度とします。
設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、XV-c1.iniファイルにキーワードを記述することで行います。
XV-c1.iniファイル
CRYSTAL_SEARCH CSP_SPGP=(P21/C) CSP_RSTEP=20.0
CONFLEXで結晶構造探索を行う場合、「CRYSTAL_SEARCH」キーワードを記述します。
探索に利用する空間群は、「CSP_SPGP=」キーワードで設定します。ここでは、P21/cを指定します。
XV-c1.molとXV-c1.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par XV-c1enter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
計算が終了すると、探索計算の詳細情報が出力されたXV-c1.cspファイルが得られます。
XV-c1.cspファイル内の「*** PREDICTED CRYSTAL STRUCTURES:」部には、結晶構造探索計算で見つかった2-amino-4-methylpyrimidineと2-methylbenzoic acidの共結晶構造に関する情報が結晶エネルギー順で出力されます。
各構造のデータはXV-c1-PCS.cifファイルに出力されています。
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
46 19 1 -112.2561 -73.1657 -39.0905 1384.6750 1.1758 6.3597 14.7215 17.2181 90.0000 120.8001 90.0000 P21/C 426 7035 20.00 0
54 487 2 -112.1447 -72.8870 -39.2577 1330.3934 1.2238 7.4330 26.1512 7.7427 90.0000 62.1242 90.0000 P21/C 436 7188 20.00 0
55 6106 3 -112.1190 -73.0217 -39.0973 1373.5185 1.1854 7.2525 14.8932 15.7678 90.0000 53.7521 90.0000 P21/C 421 6951 20.00 0
57 1692 4 -112.0257 -72.0386 -39.9871 1368.6700 1.1895 8.9646 9.9375 27.1501 90.0000 145.5370 90.0000 P21/C 431 7128 20.00 0
66 3542 5 -111.8336 -73.1270 -38.7065 1409.7885 1.1549 13.5655 12.0776 27.5867 90.0000 18.1748 90.0000 P21/C 421 6948 20.00 0
81 1208 6 -111.7751 -73.3963 -38.3788 1415.1006 1.1505 13.0894 10.0679 11.9525 90.0000 63.9490 90.0000 P21/C 430 7092 20.00 0
109 33 7 -111.7587 -73.2931 -38.4656 1403.7919 1.1598 26.0879 11.1575 8.6903 90.0000 146.2923 90.0000 P21/C 420 6936 20.00 0
134 4315 8 -111.7046 -72.2275 -39.4771 1399.2931 1.1635 7.1345 12.0828 25.4413 90.0000 39.6451 90.0000 P21/C 426 7035 20.00 0
140 1769 9 -111.5333 -72.9261 -38.6073 1414.9373 1.1507 12.3562 8.0869 14.2635 90.0000 96.8972 90.0000 P21/C 412 6807 20.00 0
141 967 10 -111.5221 -72.4012 -39.1210 1351.3577 1.2048 9.9354 10.2689 13.7854 90.0000 106.0926 90.0000 P21/C 432 7152 20.00 0
ここで、実験構造(左図)とエネルギー順位3の予測結晶構造(右図)とを比較すると、両者がよく一致することが分かります。
【PXRDデータを利用した結晶構造探索】
粉末X線結晶構造解析による結晶構造決定に結晶構造探索計算を応用する方法を説明します。
ここでは、Haynesらが報告したパモ酸の結晶構造(D. A. Haynes et al., Acta Cryst. 2006, E62, o1170.)を例として取り上げます。まず、パモ酸の分子構造ファイルを作成します。分子構造ファイルの作成には、PerkinElmer社のChemDrawを用います。ChemDrawの利用方法は、マニュアルをご覧ください。
パモ酸の分子構造は、MDL-MOL形式で、“pamoic-acid.mol”として保存します。
pamoic-acid.molファイル
pamoic-acid.mol
ChemDraw10031813162D
29 32 0 0 0 0 0 0 0 0999 V2000
-2.1471 -0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4326 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.4326 0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.1471 0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 -0.4125 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5761 -0.8250 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5761 -1.6500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 -2.0625 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.1471 -1.6500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.4294 -1.0931 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7182 -0.4538 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 -0.8662 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1471 -0.4538 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1471 0.3712 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 0.7837 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7182 0.3712 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 1.6087 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.7182 2.0212 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0037 1.6087 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.0037 0.7837 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.4326 -1.6912 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8616 -0.8663 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
3.5761 -0.4538 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8616 -1.6913 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.1471 1.6500 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.8616 2.0625 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.4326 2.0625 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.7182 0.8250 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
1 2 2 0
2 3 1 0
3 4 2 0
4 5 1 0
5 6 2 0
6 1 1 0
6 7 1 0
7 8 2 0
8 9 1 0
9 10 2 0
10 1 1 0
2 11 1 0
11 12 1 0
12 13 1 0
13 14 2 0
14 15 1 0
15 16 2 0
16 17 1 0
17 12 2 0
16 18 1 0
18 19 2 0
19 20 1 0
20 21 2 0
21 17 1 0
13 22 1 0
14 23 1 0
23 24 2 0
23 25 1 0
4 26 1 0
26 27 2 0
26 28 1 0
3 29 1 0
M END
次に、パモ酸の分子構造最適化を行います。
[Interfaceから実行する場合]
pamoic-acid.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。孤立分子の構造最適化が始まります。
[コマンドラインから実行する場合]
pamoic-acid.molをフォルダに格納し、下記コマンドを実行してください。計算が始まります。なお、iniファイルがない場合、CONFLEXは孤立分子の構造最適化をデフォルト設定にて行います。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par pamoic-acidenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
次に、パモ酸の配座探索を行います。
[Interfaceから実行する場合]
pamoic-acid-F.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログの「Calculation Type:」のプルダウンメニューから「Confromation Search」を選択します。「Search Limit:」の値は、「30.0」とします。これは、配座探索空間の範囲を決めます。
設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、pamoic-acid-F.iniファイルにキーワードを記述することで行います。
pamoic-acid-F.iniファイル
CONFLEX SEL=30.0
CONFLEXで配座探索を行う場合、「CONFLEXL」キーワードを記述します。
「SEL=30.0」は、配座探索に使用する初期構造のエネルギー範囲(Search Limit)を30.0 kcal/molとすることを意味します。
pamoic-acid-F.molとpamoic-acid-F.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par pamoic-acid-Fenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
配座探索の結果、118の配座異性体が得られます。ここでは、3番目にエネルギーの低い配座異性体を結晶構造探索計算に用います。そこで、pamoic-acid-F.sdfファイルから、3番目にエネルギーの低い配座異性体の分子構造ファイルを“pamoic-acid-c3.mol”として作成します。pamoic-acid-F.sdfファイルは、pamoic-acid-F.molファイル等と同じフォルダに格納されています。
pamoic-acid-c3.molファイル
pamoic-acid.mol
CONFLEX 18102311013D 1 1.00000 57.28062 16
C2 ,E = 57.281, G = 0.144E-07, P = 4.5793, M( 0), IFN =00000003-00000016
45 48 0 0 999 V2000
-1.3167 2.0796 -0.8640 C 0 0 0 0 0
-0.1166 1.2932 -0.8876 C 0 0 0 0 0
1.0079 1.7433 -0.1606 C 0 0 0 0 0
0.9563 2.9081 0.6175 C 0 0 0 0 0
-0.2214 3.6554 0.6552 C 0 0 0 0 0
-1.3466 3.2641 -0.0791 C 0 0 0 0 0
-2.5025 4.0623 -0.0282 C 0 0 0 0 0
-3.6457 3.7218 -0.7444 C 0 0 0 0 0
-3.6457 2.5743 -1.5200 C 0 0 0 0 0
-2.5025 1.7708 -1.5777 C 0 0 0 0 0
-0.0000 0.0000 -1.6979 C 0 0 0 0 0
0.1166 -1.2932 -0.8876 C 0 0 0 0 0
-1.0079 -1.7433 -0.1606 C 0 0 0 0 0
-0.9563 -2.9081 0.6175 C 0 0 0 0 0
0.2214 -3.6554 0.6552 C 0 0 0 0 0
1.3466 -3.2641 -0.0791 C 0 0 0 0 0
1.3167 -2.0796 -0.8640 C 0 0 0 0 0
2.5025 -4.0623 -0.0282 C 0 0 0 0 0
3.6457 -3.7218 -0.7444 C 0 0 0 0 0
3.6457 -2.5743 -1.5200 C 0 0 0 0 0
2.5025 -1.7708 -1.5777 C 0 0 0 0 0
-2.1663 -1.0131 -0.2688 O 0 0 0 0 0
-2.1487 -3.3401 1.3832 C 0 0 0 0 0
-3.2090 -2.7368 1.4055 O 0 0 0 0 0
-1.9746 -4.4795 2.0733 O 0 0 0 0 0
2.1487 3.3401 1.3832 C 0 0 0 0 0
3.2090 2.7368 1.4055 O 0 0 0 0 0
1.9746 4.4795 2.0733 O 0 0 0 0 0
2.1663 1.0131 -0.2688 O 0 0 0 0 0
-0.2634 4.5620 1.2557 H 0 0 0 0 0
-2.5220 4.9676 0.5759 H 0 0 0 0 0
-4.5311 4.3490 -0.6932 H 0 0 0 0 0
-4.5334 2.2918 -2.0797 H 0 0 0 0 0
-2.5709 0.8759 -2.1893 H 0 0 0 0 0
-0.8381 -0.1310 -2.3887 H 0 0 0 0 0
0.8381 0.1310 -2.3887 H 0 0 0 0 0
0.2634 -4.5620 1.2557 H 0 0 0 0 0
2.5220 -4.9676 0.5759 H 0 0 0 0 0
4.5311 -4.3490 -0.6932 H 0 0 0 0 0
4.5334 -2.2918 -2.0797 H 0 0 0 0 0
2.5709 -0.8759 -2.1893 H 0 0 0 0 0
-2.8489 -1.3798 0.3330 H 0 0 0 0 0
-2.8335 -4.6307 2.5208 H 0 0 0 0 0
2.8335 4.6307 2.5208 H 0 0 0 0 0
2.8489 1.3798 0.3330 H 0 0 0 0 0
1 2 2 0 0
2 3 1 0 0
3 4 2 0 0
4 5 1 0 0
5 6 2 0 0
6 1 1 0 0
6 7 1 0 0
7 8 2 0 0
8 9 1 0 0
9 10 2 0 0
10 1 1 0 0
2 11 1 0 0
11 12 1 0 0
12 13 1 0 0
13 14 2 0 0
14 15 1 0 0
15 16 2 0 0
16 17 1 0 0
17 12 2 0 0
16 18 1 0 0
18 19 2 0 0
19 20 1 0 0
20 21 2 0 0
21 17 1 0 0
13 22 1 0 0
14 23 1 0 0
23 24 2 0 0
23 25 1 0 0
4 26 1 0 0
26 27 2 0 0
26 28 1 0 0
3 29 1 0 0
5 30 1 0 0
7 31 1 0 0
8 32 1 0 0
9 33 1 0 0
10 34 1 0 0
11 35 1 0 0
11 36 1 0 0
15 37 1 0 0
18 38 1 0 0
19 39 1 0 0
20 40 1 0 0
21 41 1 0 0
22 42 1 0 0
25 43 1 0 0
28 44 1 0 0
29 45 1 0 0
M END
次に、結晶構造探索計算に利用する参照回折データを作成します。
Haynesらの論文には、回折データ(cv6632Isup2.rtv)が添付されていますので、これを利用し参照回折データ(pamoic-acid-c3.xrd)を作成します。
pamoic-acid-c3.xrdファイル
Co 8.010 79.990 0.01 7199 8.010 0.000000 8.020 0.000000 8.030 0.000000 8.040 91.252560 8.050 9.827270 8.060 0.000000 (中略) 79.970 43.877350 79.980 17.738100 79.990 42.620530
1行目はX線源の元素名、2行目は2θの範囲、刻み値、および回折データの総数です。
3行目以降は、2θの値と回折強度です。回折強度には、「_pd_meas_counts_total」の値から「_pd_proc_intensity_bkg_calc」の値を引くことで、バックグラウンドを差し引いた強度データを利用していますバックグランドを差し引くことで回折強度が負の値になる場合、強度は“0”としました。
なお、xrdファイルには空行を含めないように注意してください。
pamoic-acid-c3.xrdファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\crystal\powder\pamoic-acid-c3.xrd)。
xrdファイルの1行目は、波長の値を記述することも可能です。また、参照回折データはバックグラウンドを差し引いたものを利用してください。
最後に、結晶構造探索計算を行います。
[Interfaceから実行する場合]
pamoic-acid-c3.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが開きます。
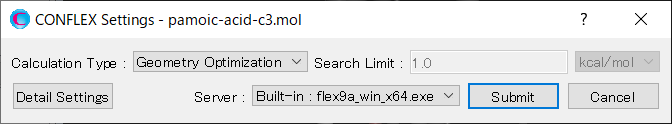
結晶構造探索計算を実施するために、詳細設定ダイアログの「General Settings」ダイアログにある「Calculation Type:」のプルダウンメニューから、「Crystal Search」を選択します。
結晶構造の最適化方法は、デフォルトで「ALL」となります。変更は、「Crystal Calculation」ダイアログの「Crystal optimization:」プルダウンメニューからできます。
また、本ダイアログでは、カットオフ距離や計算方法等の分子間相互作用計算に関する設定も可能です。
次に、「Crystal Search」ダイアログで、結晶構造探索計算の設定を行います。
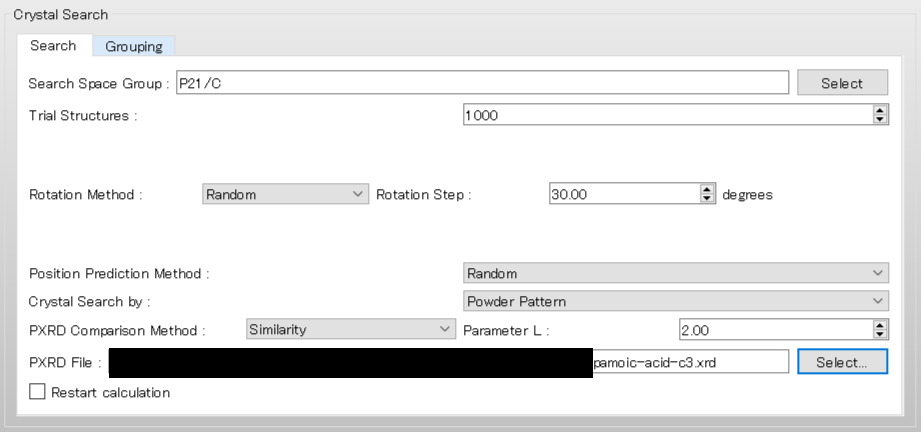
まず、探索に利用する空間群をP21/cとするため、「Search Space Group:」を「P21/C,P212121」から「P21/C」に編集します。
「Rotation Method:」と「Position Prediction Method:」は、分子の回転方法と配置方法を設定します。ここでは、ランダムに決めるとして、両者とも「Random」を選択します。
「Trial Structures:」は、生成する試行結晶構造数を設定します。ここでは、試行結晶構造数は1000とします。
次に、「Crystal Search by:」を「Powder Pattern」に変更します。これにより、探索により見つかった結晶構造は、参照回折データに基づいて評価されます。
最後に、「PXRD File:」のをクリックし、参照回折データとなるpamoic-acid-c3.xrdファイル選択します。
設定が終わりましたら、をクリックします。計算が始まります。
[コマンドラインから実行する場合]
計算設定は、pamoic-acid-c3.iniファイルにキーワードを記述することで行います。
pamoic-acid-c3.iniファイル
CRYSTAL_SEARCH CSP_SEARCH=POWDER_PATTERN CSP_SPGP=(P21/C) CSP_AUS_MODE=RANDOM CSP_ROT_MODE=RANDOM CSP_MAX_CRYSTAL=1000 CRYSTAL_OPTIMIZATION=ALL
CONFLEXで結晶構造探索を行う場合、「CRYSTAL_SEARCH」を記述します。
「CSP_SEARCH=POWDER_PATTERN」は、探索計算より見つかった結晶構造を、参照回折データに基づいて評価することを意味します。
「CSP_SPGP=P21/C」は、探索計算に空間群P21/cを利用することを意味します。
「CSP_ROT_MODE=RANDOM」と「CSP_AUS_MODE=RANDOM」は、それぞれ分子の回転と配置をランダムに決めることを示します。
「CSP_MAX_CRYSTAL=1000」は、生成する試行結晶構造数を1000に設定しています。
「CRYSTAL_OPTIMIZATION=ALL」は、最適化方法として「ALL」を設定し、非対称単位の分子構造、分子配向、分子位置、および格子定数を最適化することを意味します。
pamoic-acid-c3.molとpamoic-acid-c3.ini、pamoic-acid-c3.xrdの三つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par pamoic-acid-c3enter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
計算が終了すると、探索計算の詳細情報が出力されたpamoic-acid-c3.cspファイルが得られます。
参照回折データを用いた結晶構造探索では、探索計算で見つかった結晶構造は、それら結晶構造から計算される回折パターンと参照回折パターンとの類似度に基づいて評価されます。pamoic-acid-c3.cspファイル内の「*** PREDICTED CRYSTAL STRUCTURES:」部には、結晶構造探索計算で見つかったパモ酸の結晶多形構造に関する情報が類似度の高い順に出力されます。つまり順位1位の結晶構造は、参照回折パターンと最も類似した回折パターンを示す結晶構造となります。
各構造のデータはpamoic-acid-c3-PCS.cifファイルに出力されています。なお、類似度の計算はGELDER法(R. de Gelder et al, J Comput Chem 22: 273–289, 2001.)を採用しています。
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK R_RNK PXRD_SIM CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
5 19 1 1 0.9056 12.3774 59.3730 -46.9957 1829.5937 1.4089 20.3676 4.9453 19.2844 90.0000 70.3767 90.0000 P21/C 225 10125 20.00 0
9 483 135 2 0.8351 24.0764 58.4811 -34.4047 2074.3915 1.2427 4.8375 19.5849 22.8573 90.0000 106.6807 90.0000 P21/C 188 8460 20.00 0
10 260 200 3 0.8276 25.8085 59.1800 -33.3715 1913.6902 1.3470 5.0699 25.0663 16.1916 90.0000 111.5615 90.0000 P21/C 204 9180 20.00 0
12 56 69 4 0.8235 21.4836 61.1121 -39.6285 1929.2047 1.3362 12.0076 12.5142 16.0878 90.0000 127.0568 90.0000 P21/C 205 9225 20.00 0
13 322 3 5 0.8219 14.2830 58.8049 -44.5220 1897.8000 1.3583 10.1471 4.8134 39.0029 90.0000 85.0212 90.0000 P21/C 202 9090 20.00 0
17 367 89 6 0.8182 22.4699 61.5496 -39.0797 1934.6148 1.3325 10.3329 20.6152 9.1642 90.0000 82.3239 90.0000 P21/C 197 8865 20.00 0
18 60 280 7 0.8177 28.4589 58.0370 -29.5781 1981.9578 1.3006 4.7838 11.1176 37.2683 90.0000 90.6615 90.0000 P21/C 202 9090 20.00 0
19 541 143 8 0.8137 24.3246 58.3375 -34.0128 1917.4130 1.3444 4.9060 15.6069 33.7231 90.0000 47.9523 90.0000 P21/C 202 9090 20.00 0
23 20 9 9 0.8133 15.9937 59.4918 -43.4981 1888.4289 1.3650 10.3216 9.1532 22.6717 90.0000 61.8423 90.0000 P21/C 197 8865 20.00 0
24 5 45 10 0.8122 20.3188 58.3402 -38.0214 1945.9817 1.3247 4.8345 10.1697 39.5911 90.0000 88.6467 90.0000 P21/C 204 9180 20.00 0
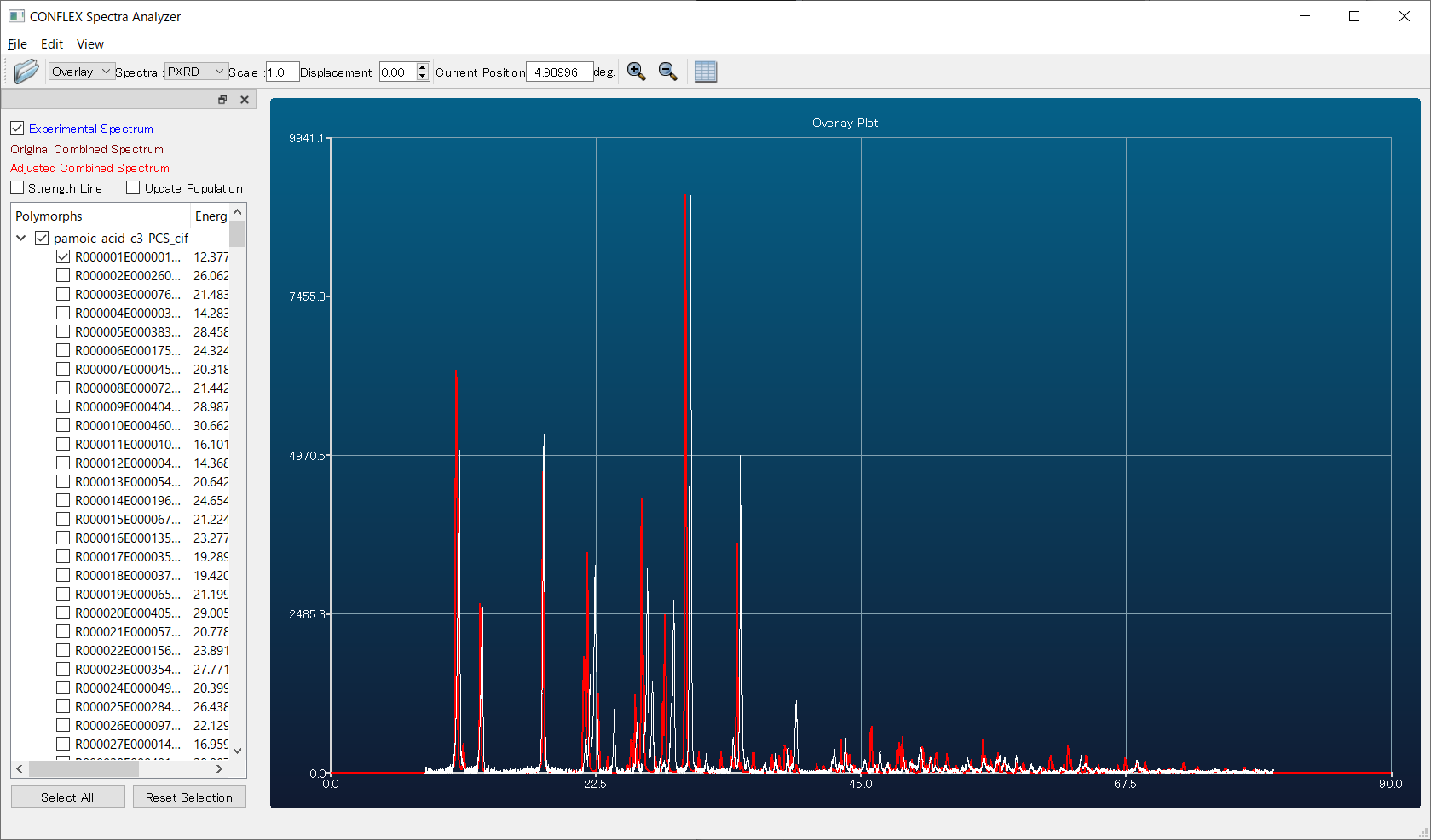
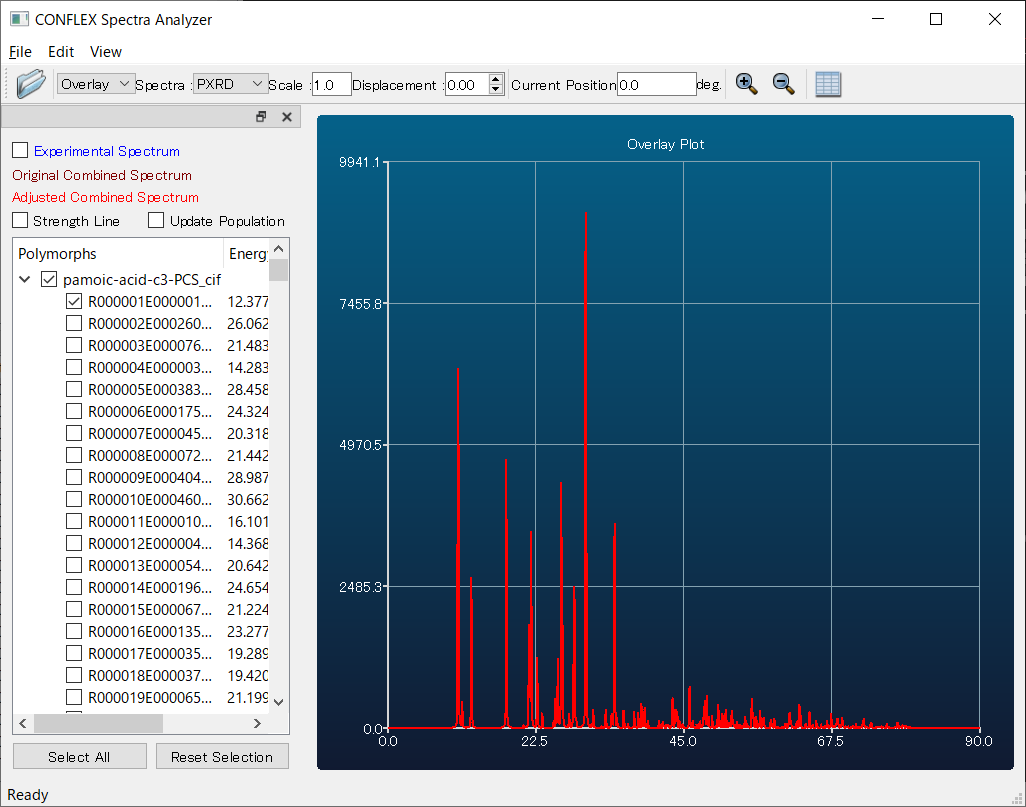
エネルギー順位1位、回折パターン類似度順位1位の結晶構造の回折パターン(赤)と実験回折パターン(白)を比較すると(下図)、回折ピーク位置や回折ピークの相対的な強度が、おおむね一致していることが分かります。リートベルト解析による構造精密化の初期構造として本構造を利用することで、真値にすばやく到達すると期待できます。
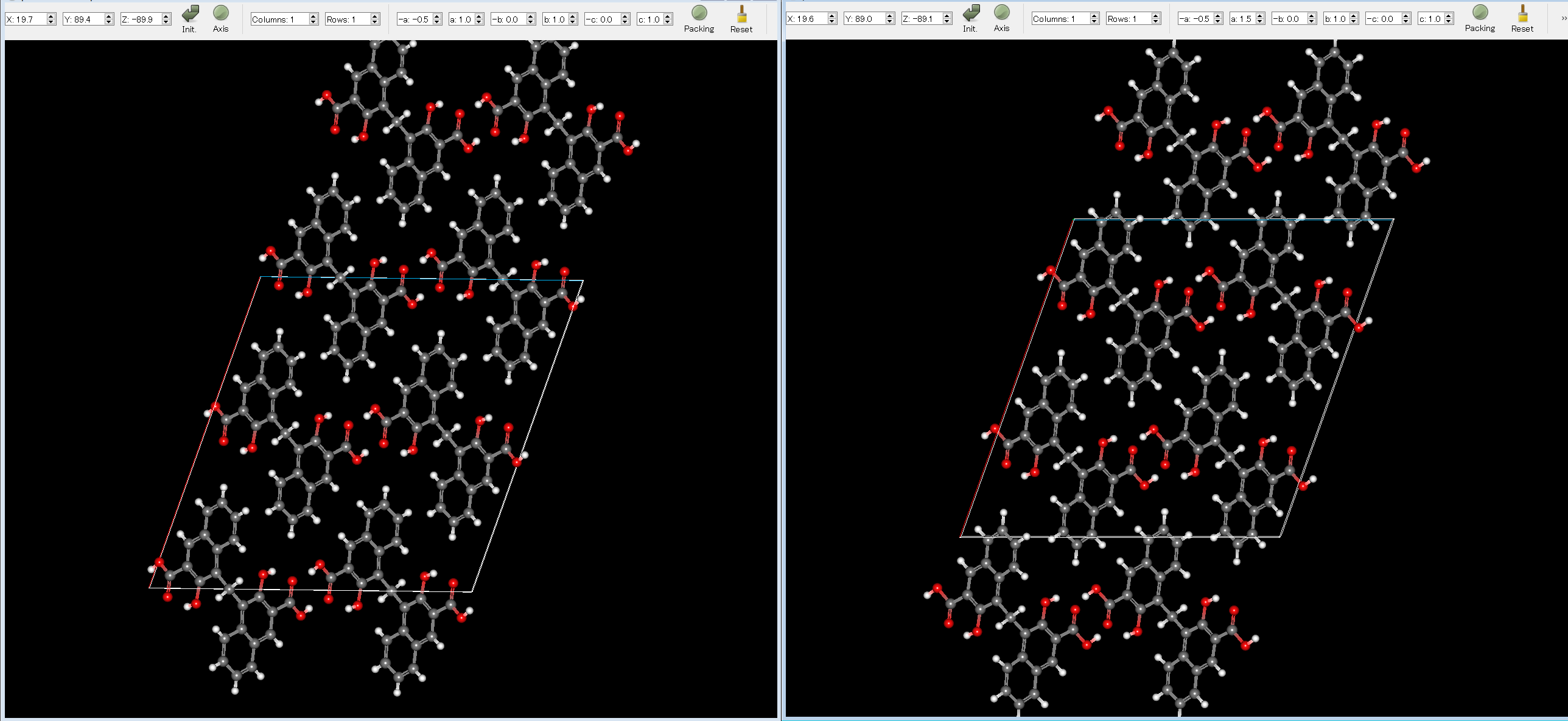
最後に、実験構造(左図)と順位1位の計算構造(右図)を示します。両構造がよく一致することがわかります。
【探索計算の出力ファイル】
結晶構造探索計算の終了後、以下のファイルが出力されます。
| ファイルの種類 | 説明 |
|---|---|
| (入力ファイル名).csp | 結晶構造探索計算に関する詳細情報が出力されます。 |
| (入力ファイル名).cpt | 再計算用の情報が出力されます。 |
| (入力ファイル名).ical | 探索で得られた結晶構造(-PCS.cifに出力された構造)の回折データが出力されます。 |
| (入力ファイル名)-PCS.cif | 探索で得られた結晶構造が結晶エネルギー順(あるいは、回折パターンの類似度順)に出力されます。 |
| (入力ファイル名)-FCS.cif | 最適化計算の終了した構造が、順に出力されます。そのため、探索計算の途中でも、本ファイルを用いることで、探索により得られた結晶構造を確認することができます。 |
cspファイルの「*** PREDICTED CRYSTAL STRUCTURES:」部分には、結晶構造探索計算から予測された結晶構造に関する情報が出力されています。結晶エネルギーに基づいて結晶構造探索を行った場合、それらの構造情報は結晶エネルギー順に並べられ出力されます。
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
4 318 1 -15.7921 4.6942 -20.4864 607.6657 1.3663 9.5996 8.3419 10.7545 90.0000 44.8779 90.0000 P21/C 365 4015 20.00 0
30 5295 2 -15.6634 4.7074 -20.3708 306.9963 1.3522 13.4774 7.0951 4.3469 90.0000 132.3915 90.0000 P21 371 4081 20.00 0
147 3622 3 -15.6125 4.7219 -20.3344 614.2783 1.3515 4.3718 19.8181 7.0899 90.0000 90.0000 90.0000 P212121 373 4103 20.00 0
167 3853 4 -15.6061 4.6925 -20.2986 612.8432 1.3547 9.6310 8.3434 7.6267 90.0000 90.0000 90.0000 P212121 325 3575 20.00 0
178 51 5 -15.5986 4.7162 -20.3147 614.9313 1.3501 4.3234 7.1069 21.9413 90.0000 114.1992 90.0000 P21/C 371 4081 20.00 0
190 90 6 -15.4618 4.7170 -20.1789 618.7977 1.3417 4.3746 7.0720 24.0800 90.0000 123.8359 90.0000 P21/C 374 4114 20.00 0
195 1948 7 -15.3029 4.7145 -20.0173 1222.4436 1.3583 14.5999 8.3633 10.4384 90.0000 73.5604 90.0000 C2/C 357 3927 20.00 0
206 169 8 -15.2948 4.7210 -20.0158 612.9536 1.3545 4.0230 8.3785 19.4984 90.0000 111.1490 90.0000 P21/C 358 3938 20.00 0
229 18 9 -15.2940 4.7263 -20.0203 612.5302 1.3554 8.3759 18.2972 8.8552 90.0000 26.8303 90.0000 P21/C 365 4015 20.00 0
290 4 10 -15.2939 4.7218 -20.0156 612.7425 1.3549 8.3793 18.2092 9.2919 90.0000 25.6065 90.0000 P21/C 357 3927 20.00 0
一方、参照回折データを用いて結晶構造探索を行った場合、それらの構造情報は、回折パターンの類似度の高い順に並べられ出力されます。
*** PREDICTED CRYSTAL STRUCTURES:
IDX CID E_RNK R_RNK PXRD_SIM CRYST INTRA INTER VOL DES A B C ALPHA BETA GAMMA SPGP NCALMOL NCALATM DMAX NNEV
5 19 1 1 0.9056 12.3774 59.3730 -46.9957 1829.5937 1.4089 20.3676 4.9453 19.2844 90.0000 70.3767 90.0000 P21/C 225 10125 20.00 0
9 483 135 2 0.8351 24.0764 58.4811 -34.4047 2074.3915 1.2427 4.8375 19.5849 22.8573 90.0000 106.6807 90.0000 P21/C 188 8460 20.00 0
10 260 200 3 0.8276 25.8085 59.1800 -33.3715 1913.6902 1.3470 5.0699 25.0663 16.1916 90.0000 111.5615 90.0000 P21/C 204 9180 20.00 0
12 56 69 4 0.8235 21.4836 61.1121 -39.6285 1929.2047 1.3362 12.0076 12.5142 16.0878 90.0000 127.0568 90.0000 P21/C 205 9225 20.00 0
13 322 3 5 0.8219 14.2830 58.8049 -44.5220 1897.8000 1.3583 10.1471 4.8134 39.0029 90.0000 85.0212 90.0000 P21/C 202 9090 20.00 0
17 367 89 6 0.8182 22.4699 61.5496 -39.0797 1934.6148 1.3325 10.3329 20.6152 9.1642 90.0000 82.3239 90.0000 P21/C 197 8865 20.00 0
18 60 280 7 0.8177 28.4589 58.0370 -29.5781 1981.9578 1.3006 4.7838 11.1176 37.2683 90.0000 90.6615 90.0000 P21/C 202 9090 20.00 0
19 541 143 8 0.8137 24.3246 58.3375 -34.0128 1917.4130 1.3444 4.9060 15.6069 33.7231 90.0000 47.9523 90.0000 P21/C 202 9090 20.00 0
23 20 9 9 0.8133 15.9937 59.4918 -43.4981 1888.4289 1.3650 10.3216 9.1532 22.6717 90.0000 61.8423 90.0000 P21/C 197 8865 20.00 0
24 5 45 10 0.8122 20.3188 58.3402 -38.0214 1945.9817 1.3247 4.8345 10.1697 39.5911 90.0000 88.6467 90.0000 P21/C 204 9180 20.00 0
「*** PREDICTED CRYSTAL STRUCTURES:」部分の各ヘッダーの意味は、下記のとおりです。
| ヘッダー名 | 説明 |
|---|---|
| E_RNK | 結晶エネルギーの順位を示します。 |
| R_RNK | 回折パターンの類似度の順位を示します。 |
| CRYST | Ecrystalを示します。 |
| INTRZA | Eintraを示します。 |
| INTER | Elatticeを示します。 |
| VOL | 単位格子の体積を示します。 |
| DES | 単位格子の密度を示します。 |
| A,B,C,ALPHA,BETA,GAMMA | 格子定数を示します。 |
| SPGP | 空間群を示します。 |
| NCALMOL | 計算に含まれる分子数を示します。 |
| NCALATM | 計算に含まれる原子数を示します。 |
| DMAX | カットオフ距離を示します。 |
| NNEV | 負の固有値の数を示します。 |
*「NCALMOL, NCALATM, DMAX」について、vdW相互作用と静電相互作用で異なるカットオフ距離を利用している場合は、vdW相互作用に関する値が表示されます。
icalファイルには、探索で得られた結晶構造(-PCS.cifに出力された構造)の回折データが出力されます。
-PCS.cifファイル内の「data_」と、icalファイル内の「NAME:」の値は、同じ結晶構造で一致します。
------------ SIMULATED POWDER PATTERNS ------------
CID: 882
NAME: R000001E000001C000882
X-RAY: CO (KA1)
WAVE: 1.78899600
2*THETA: 8.010 - 79.990 , 0.010 STEP
H K L 2*THETA INTENSITY d
(DEGREE) (ANGSTROME)
0 0 0 8.010 1.167 0.00000
0 0 0 8.020 1.174 0.00000
0 0 0 8.030 1.182 0.00000
0 0 0 8.040 1.190 0.00000
0 0 0 8.050 1.197 0.00000
0 0 0 8.060 1.205 0.00000
0 0 0 8.070 1.213 0.00000
0 0 0 8.080 1.221 0.00000
0 0 0 8.090 1.229 0.00000
0 0 0 8.100 1.237 0.00000
0 0 0 8.110 1.246 0.00000
0 0 0 8.120 1.254 0.00000
0 0 0 8.130 1.263 0.00000
0 0 0 8.140 1.271 0.00000
(以下略)
【探索結果の可視化】
結晶構造探索計算により得られた結晶多形構造は、「(入力ファイル名)-PCS.cif」ファイルに出力されています。これを、CONFLEX Interfaceにて開くことで、得られた構造を可視化することができます。
また、(入力ファイル名)-PCS.cifファイルを開いた状態で、「Application」メニューから「Spectra_Analyzer」を選択することで、各構造の回折パターンを可視化させることができます。
【計算の再実行】
[Interfaceから実行した場合]
計算に利用した分子構造ファイルとiniファイル、計算後に出力されたcptファイルを同じフォルダに格納します。参照回折データを用いた探索計算の場合は、xrdファイルも必要です。
次に、分子構造ファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが開きます。
結晶構造探索計算を再実行するために、詳細設定ダイアログの「Crystal Search」ダイアログの下部にある「Restart calculation」チェックボックスにチェックを入れます。
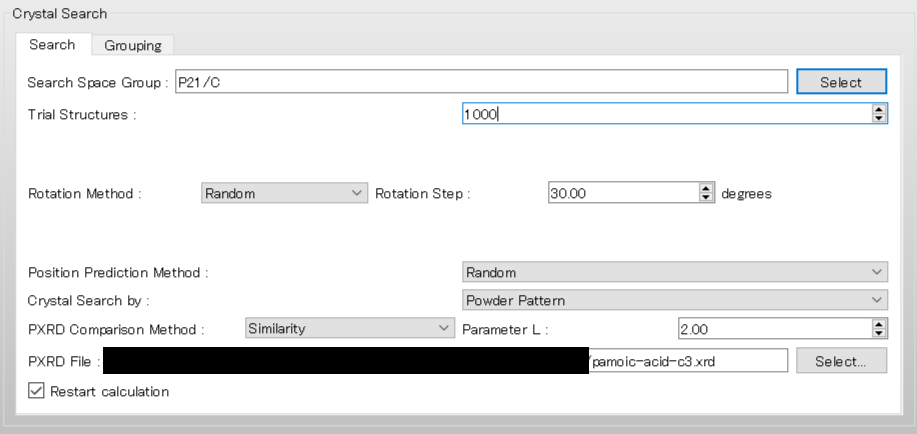
その他の設定は、iniファイルの内容から自動に設定されています。をクリックしてください。
もし、再実行させる計算を「から実行し、手動にてキーワードを追加している場合は、同じキーワードを追加してください。探索に関する設定が一致していないと、正しく再実行ができない場合があります。
[コマンドラインから実行した場合]
計算に利用した分子構造ファイルとiniファイル、計算後に出力されたcptファイルを同じフォルダに格納します。参照回折データを用いた探索計算の場合は、xrdファイルも必要です。
次に、cptファイルの拡張子をrstに変更してください。また、iniファイル内に「CSP_RESTART」キーワードを追加してください。探索に関する設定は、変更しないように注意してください。設定が一致していないと、正しく再実行ができない場合があります。
入力ファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par (入力ファイル名)enter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
【利用可能な空間群】
230種の空間群のうち、次に示す空間群以外は利用できます。
P4MM, P4BM, P42CM, P42NM, I4MM, I4CM, P-42M, P-421M, I-42M, P4/MMM, P4/NBM, P4/MBM,
P4/NMM, P42/MCM, P42/NNM, P42/MNM, P42/NCM, I4MMM, I4/MCM, P3, P31, P32, R3, P-3,
R-3, P312, P321, P3112, P3121, P3212, P3221, R32, P3M1, P31M, P3C1, P31C, R3M, R3C,
P-31M, P-31C, P-3M1, P-3C1, R-3M, R-3C, P6, P61, P65, P62, P64, P63, P-6, P6/M,
P63/M, P622, P6122, P6522, P6222, P6422, P6322, P6MM, P6CC, P63CM, P63MC, P-6M2,
P-6C2, P-62M, P-62C, P6/MMM, P6/MCC, P63/MCM, P63/MMC, P23, F23, I23, P213, I213,
PM-3, PN-3, FM-3, FD-3, IM-3, PA-3, IA-3, P432, P4232, F432, F4132, I432, P4332,
P4132, I4132, P-43M, F-43M, I-43M, P-43N, F-43C, I-43D, PM-3M, PN-3N, PM-3N, PN-3M,
FM-3M, FM-3C, FD-3M, FD-3C, IM-3M, IA-3D