【構造最適化と振動解析】
CONFLEXによる分子の構造最適化と振動解析について説明します。
【構造最適化と振動解析の実行】
Cyclohexaneを例として、構造最適化と振動解析を実行する方法について説明します。まず、Cyclohexaneの分子構造データをMDL-MOL形式で、Cyclohexane.molとして準備します。
Cyclohexane.molファイルはCONFLEXのインストールフォルダ内のSample_Filesフォルダにあります(Sample_Files\CONFLEX\optimization_and_search\Cyclohexane.mol)。
Cyclohexane.molファイル

- A1:原子の数(999まで)
- A2:結合の数(999まで)
- A3:現在使用されていません
- A4:現在使用されていません
- A5:光学活性中心の存在(0:光学活性中心なし、1:光学活性中心あり):現在使用されていません
- B1:原子のデカルト座標と原子記号
- B2:原子の特性リスト(同位体、電荷、立体中心等を示します。通常、電荷と立体中心しか必要としません。)
- C1:原子同士の結合情報(ある原子を中心とした場合の隣接する原子の結合情報を示します。結合状態、立体状態、結合トポロジー等を示します。通常は、隣接原子間の結合情報、結合状態、立体状態しか必要としません。)
- D1:原子座標の終了であることを示し、必須情報です。
[Interfaceから実行する場合]
Cyclohexane.molファイルをCONFLEX Interfaceを用いて開きます。

Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。分子の構造最適化計算と振動計算が始まります。

[コマンドラインから実行する場合]
Cyclohexane.molをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Cyclohexaneenter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
なお、計算設定ファイルであるiniファイルがない場合、CONFLEXは分子の構造最適化計算と振動計算をデフォルト設定にて行います。
この場合、Cyclohexane.iniファイルに「MMFF94S OPT=NEWTON OPTBY=ENERGY」と指定したときと同じ計算となります。
【構造最適化計算の出力ファイル】
計算が正常に終了すると、以下の3つのファイルが保存されます。
Cyclohexane-F.mol
最適化により得られた構造を、入力ファイルと同じMDL-MOL形式で出力したファイルです。
Cyclohexane.mol
CONFLEX 20090414283D 1 1.00000 -3.56094 0
D3D ,E = -3.561, G = 6.035E-11, M(0) MMFF94S(2010-12-04HG)
18 18 0 0 999 V2000
-1.2627 0.7290 0.2258 C 0 0 0 0 0
0.0000 1.4580 -0.2258 C 0 0 0 0 0
-1.2627 -0.7290 -0.2258 C 0 0 0 0 0
-2.1462 1.2391 -0.1742 H 0 0 0 0 0
-1.3365 0.7716 1.3195 H 0 0 0 0 0
-0.0000 2.4782 0.1742 H 0 0 0 0 0
0.0000 1.5433 -1.3195 H 0 0 0 0 0
0.0000 -1.4580 0.2258 C 0 0 0 0 0
-2.1462 -1.2391 0.1742 H 0 0 0 0 0
-1.3365 -0.7716 -1.3195 H 0 0 0 0 0
0.0000 -2.4782 -0.1742 H 0 0 0 0 0
0.0000 -1.5433 1.3195 H 0 0 0 0 0
1.2627 -0.7290 -0.2258 C 0 0 0 0 0
2.1462 -1.2391 0.1742 H 0 0 0 0 0
1.3365 -0.7716 -1.3195 H 0 0 0 0 0
1.2627 0.7290 0.2258 C 0 0 0 0 0
1.3365 0.7716 1.3195 H 0 0 0 0 0
2.1462 1.2391 -0.1742 H 0 0 0 0 0
2 1 1 0 0
3 1 1 0 0
1 4 1 0 0
1 5 1 0 0
2 6 1 0 0
2 7 1 0 0
2 16 1 0 0
3 8 1 0 0
3 9 1 0 0
3 10 1 0 0
11 8 1 0 0
12 8 1 0 0
13 8 1 0 0
14 13 1 0 0
13 15 1 0 0
16 13 1 0 0
16 17 1 0 0
16 18 1 0 0
M END
Cyclohexane.bsf
最適化構造の座標やエネルギー値などを出力したファイルです。
999.9 6 0 6 0 1 0 010.00 1.00E-06 1.00E-06 0 0 25.000010001100000000000 Cyclohexane: Cyclohexane.mol D3D ,E = -3.561, G = 6.035E-11, M(0) MMFF94S(2010-12-04HG) 18 18 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 0.00000 -1.262673724 0.729005014 0.225782342 6 0 12 2 3 4 5 0 0 1 0.000000000 1.458010029 -0.225782342 6 0 12 1 6 7 16 0 0 2 -1.262673724 -0.729005014 -0.225782342 6 0 12 1 8 9 10 0 0 3 -2.146189104 1.239102857 -0.174187977 1 0 1 1 0 0 0 0 0 4 -1.336507200 0.771632792 1.319462875 1 0 1 1 0 0 0 0 0 5 -0.000000000 2.478205714 0.174187977 1 0 1 2 0 0 0 0 0 6 0.000000000 1.543265583 -1.319462875 1 0 1 2 0 0 0 0 0 7 0.000000000 -1.458010029 0.225782342 6 0 12 3 11 12 13 0 0 8 -2.146189104 -1.239102857 0.174187977 1 0 1 3 0 0 0 0 0 9 -1.336507200 -0.771632792 -1.319462875 1 0 1 3 0 0 0 0 0 10 0.000000000 -2.478205714 -0.174187977 1 0 1 8 0 0 0 0 0 11 0.000000000 -1.543265583 1.319462875 1 0 1 8 0 0 0 0 0 12 1.262673724 -0.729005014 -0.225782342 6 0 12 8 14 15 16 0 0 13 2.146189104 -1.239102857 0.174187977 1 0 1 13 0 0 0 0 0 14 1.336507200 -0.771632792 -1.319462875 1 0 1 13 0 0 0 0 0 15 1.262673724 0.729005014 0.225782342 6 0 12 2 13 17 18 0 0 16 1.336507200 0.771632792 1.319462875 1 0 1 16 0 0 0 0 0 17 2.146189104 1.239102857 -0.174187977 1 0 1 16 0 0 0 0 0 18
Cyclohexane.bso
計算に用いた力場パラメーターの数値や、各相互作用のエネルギー、更に基準振動解析により得られた熱力学的諸量、振動数、振動モードを出力したファイルです。

- (A-1):選択された力場パラメーター
- (A-2):選択された力場における分子構造に含まれる原子タイプ情報
- (A-3):選択された力場における伸縮相互作用項の関数と使用されるパラメーターに関する情報
- 以下「ANGLES:」等、各相互作用項の関数とパラメーターに関する情報が出力されます。

- (A-4):入力(初期)構造の座標と原子番号
- (A-5):自動的に判定された原子タイプ
- (A-6):自動判定された結合表
- (A-7):入力(初期)構造のエネルギー勾配

- (A-8):入力(初期)構造の慣性モーメントと双極子モーメント
- (A-9):入力(初期)構造の各相互作用関数項の立体エネルギー

- (A-10):Newton-Raphson法による構造最適化の経過
- (A-11):構造最適化が収束した
- (A-12):最終固有値の一覧。Cut-offの絶対値以下の固有値を0とすると、直線分子の場合は5個、それ以外では6個の固有値が0。

- (A-13): 計算結果の概略を表示した後に、分子構造に関する詳細な情報を選択された力場関数の相互作用項毎に表示

- (A-14):熱力学的諸関数の詳細、指定された温度、最適化構造の対称性を考慮して、内部エネルギー、エンタルピー、エントロピー、Gibbs自由エネルギー、定圧熱容量に関する分配関数量が計算される
- (A-15):虚数振動、ゼロ振動、実振動の数。6個のゼロ振動と虚数振動がないことを確認すること。

- (A-16): 振動数と振動モード。振動モードは、デカルト座標変位ベクトルとして表記される。オプション指定で内部座標系表記も可能。
【計算結果の可視化】
[Interfaceから実行した場合]
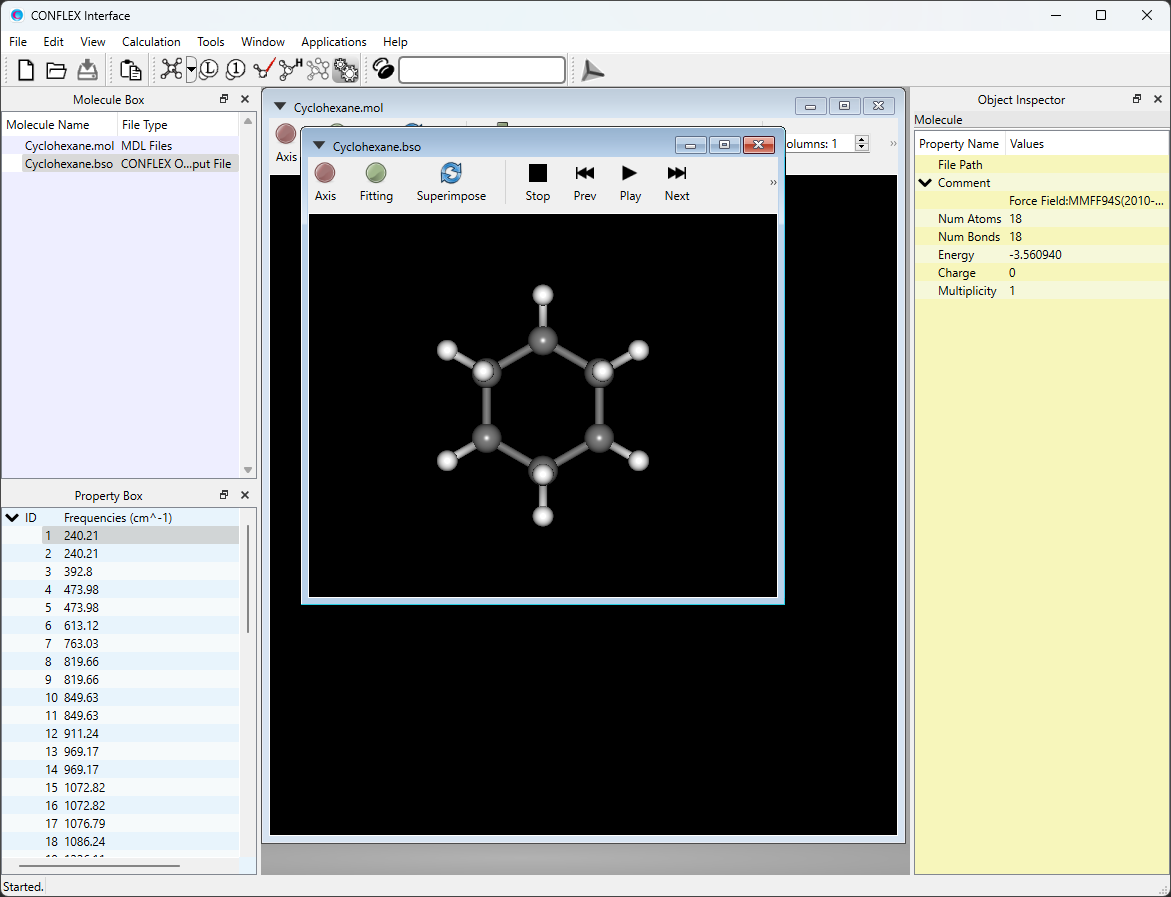
計算の実行後、CONFLEX Interfaceの下部にJob Managerが現れます。Job Managerには、実行した計算の状態が表示されます。

構造最適化を行ったJobのStateがFinishedであることを確認し、表示部分(赤枠部分)をダブルクリックしてください。
Cyclohexane.bsoが開き、最適化構造が描画されます。なお、Cyclohexane.bsoファイルは入力ファイルを格納したフォルダにあります。

Viewメニューの「Vibration」を選択すると、基準振動による原子の変位ベクトルを表示することができます。

*矢印の大きさは、Viewメニューの「Controller」を選択し表示されるツールバーから変更できます。
[コマンドラインから実行した場合]
入力ファイルを格納したフォルダに、Cyclohexane-F.molとCyclohexane.bsoファイルがあります。これらをCONFLEX Interfaceで開くことで、最適化構造を可視化できます。
Cyclohexane.bsoファイルを開いた場合、Viewメニューの「Vibration」を選択することで、基準振動による原子の変位ベクトルを表示することができます。
*矢印の大きさは、Viewメニューの「Controller」を選択し表示されるツールバーから変更できます。
【溶媒効果を取り入れた計算】
GB/SAモデルによる溶媒効果の導入
化学実験において、化合物を合成したり構造や物性値を測定したりする場合、その化合物は溶媒に溶かした状態で扱うことがほとんどです。ですから、分子計算を用いて実験により得られた現象を解析する場合、対象とする実験が化合物を溶液で扱っているのであれば、計算も溶液の状態を考慮したものでなければなりません。しかし通常の分子力場などは、気相中の化合物の構造や振動などを再現するよう構築されており、溶媒からの寄与は考慮されていません。分子シミュレーションで溶媒からの寄与を再現するためには、溶質の周りに多くの溶媒分子を配置させた計算を行えばいいのですが、そうすると今度は溶媒分子の数だけ系の自由度が増えてしまい計算時間が大幅に増加してしまいます。
この不具合を解消するため、分子計算の分野では「連続誘電体モデルによる溶媒効果の導入」が古くから研究されています。CONFLEXでは、この連続誘電体モデルの一つであり分子力学計算において最も広く用いられているGB/SAモデルを採用しており、溶媒効果を取り入れた構造最適化・振動解析、配座探索、および溶媒和自由エネルギーの算出が可能です。
なお現バージョンでは、力場はMMFF94sに、溶媒は水とオクタノールに対応しています。
GB/SAモデルとは?
GB/SAモデルとは、溶媒による静電的な寄与を一般化Born(Generalized Born, GB)式を用いて算出し、非静電的な寄与は溶媒和接触可能表面積(Solvent-Accessible Surface Area, SA)を元に算出するモデルのことです。溶媒によるエネルギーの増加分を

と表すと、静電項はGB式より

となり、また非静電項は

として算出します。ここでqiは原子i上の電荷、rijは原子i - j間距離、αijおよびDijは有効Born半径より求められる値、σkは表面張力係数、SAkは原子kの溶媒和接触可能表面積です。
尚、CONFLEX 9より、計算中この非静電項を無視するオプション「SA=IGNORE」を追加しましたので、構造最適化が収束しない場合はこちらを設定した計算をお試しください。
GB/SAモデルを導入した計算
zwitter ion型のグリシン2量体を例にします。

座標データ(gly2.mol)
gly2.mol
17 16 0 0 0 0 0 0 0 0999 V2000
-1.2219 0.9765 -2.1504 N 0 3 0 0 0 0 0 0 0 0 0 0
0.2781 0.9765 -2.1504 C 0 0 0 0 0 0 0 0 0 0 0 0
0.6496 2.0256 -2.1504 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7803 0.2658 -0.9176 C 0 0 0 0 0 0 0 0 0 0 0 0
1.9679 0.1552 -0.7258 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.1098 -0.2537 -0.0165 N 0 0 0 0 0 0 0 0 0 0 0 0
0.3728 -0.9366 1.1681 C 0 0 0 0 0 0 0 0 0 0 0 0
0.9911 -0.2372 1.7741 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.8018 -1.4089 1.9892 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6088 -2.0256 3.0592 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.9679 -1.1835 1.5993 O 0 5 0 0 0 0 0 0 0 0 0 0
-1.5708 0.4839 -3.0034 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.5698 0.4832 -1.2973 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.5701 1.9618 -2.1504 H 0 0 0 0 0 0 0 0 0 0 0 0
0.6489 0.4518 -3.0592 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.1047 -0.1611 -0.1772 H 0 0 0 0 0 0 0 0 0 0 0 0
0.9910 -1.8114 0.8658 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 12 1 0
1 13 1 0
1 14 1 0
2 3 1 0
2 4 1 0
2 15 1 0
4 5 2 0
4 6 1 0
6 7 1 0
6 16 1 0
7 8 1 0
7 9 1 0
7 17 1 0
9 10 2 0
9 11 1 0
M END
[Interfaceから実行する場合]
gly2.molファイルをCONFLEX Interfaceを用いて開きます。

Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。詳細設定ダイアログが開きます。

次に、詳細設定ダイアログの「Force Field」ダイアログにある「Solvent Effect」 のプルダウンメニューから「GBSA」を選択します。

設定が終わりましたら、詳細設定ダイアログのをクリックします。溶媒効果を含む構造最適化が行われます。
[コマンドラインから実行する場合]
計算設定は、gly2.iniファイルにキーワードを記述することで行います。
gly2.iniファイル
MMFF94S GBSA
「GBSA」のキーワードを追加することで、GB/SAモデルを導入した計算が実行できます。
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
gly2.molとgly2.iniをフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par gly2enter
上記は、Windowsの場合です。他のOSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
最後に、最適化後の構造を下記に示します。可視化の方法は、「計算結果の可視化」をご覧ください。

【構造を部分的に固定した最適化計算】
構造最適化によりエネルギー極小構造を求める場合、通常は系に含まれる全原子の自由度について最適化を行いますが、部分構造が既知でそれ以外の未知の構造についてのみ最適化したい場合や、ある特定の構造パラメーターを一定に保ったまま最適化したい場合など、系の部分構造を固定した最適化を実施したいこともあります。
CONFLEXでは、以下に示す3通りの方法で部分構造の変化に制限を課した構造最適化を行うことができます。
また、これらの制限を課して配座探索を行うことも可能です。
*調和ポテンシャルによる構造固定
原子間の距離や結合角、二面角などの構造パラメーターについて、調和ポテンシャルを加えることで構造変化に制限を課すことができます。
それぞれの構造パラメーターに対応するキーワードは以下の通りです:
| 構造パラメーター | キーワード | 説明 |
|---|---|---|
| 原子間距離 | PSEUDO_BOND=(I,J,STD,FK) | 原子I-J間について、中心距離STD (Å) および力の定数FK (kcal・mol-1・Å-2) の調和ポテンシャルを加える。 |
| PSEUDO_HALF=(I,J,STD,FK) | 原子I-J間について、中心距離|STD| (Å) および力の定数FK (kcal・mol-1・Å-2)の半調和ポテンシャル関数を加える。STD>0ではI-J間がSTDより長くなった場合、STD<0ではI-J間が|STD|より短くなった場合にエネルギー値が増加する。 | |
| 結合角 | PSEUDO_ANGL=(I,J,K,STD,FK) | I-J-K角について、中心値STD (°) および力の定数FK (kcal・mol-1・rad-2) の調和ポテンシャルを加える。 |
| 二面角 | PSEUDO_TORS=(I,J,K,L,STD,FK) | I-J-K-L二面角について、中心値STD (°) および力の定数FK (kcal・mol-1・rad-2) の調和ポテンシャルを加える。 |
計算例
n-ブタンのtrans配座の構造に対して、「PSEUDO_TORS=」キーワードを利用して、C-C-C-C二面角を150.0度に維持させた計算を行います。
n-ブタンの3次元構造

n-ブタンの座標データ(n-butane.mol)
n-butane.mol
14 13 0 0 0 0 0 0 0 0999 V2000
-1.1802 -1.5102 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
0.1388 -0.7487 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.1388 0.7487 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.1802 1.5102 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.9774 -2.6046 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.7631 -1.2415 0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.7637 -1.2413 -0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7217 -1.0175 -0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
0.7223 -1.0177 0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.7217 1.0175 0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.7223 1.0177 -0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
0.9774 2.6046 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.7631 1.2415 -0.9093 H 0 0 0 0 0 0 0 0 0 0 0 0
1.7637 1.2412 0.9088 H 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0
1 5 1 0
1 6 1 0
1 7 1 0
2 3 1 0
2 8 1 0
2 9 1 0
3 4 1 0
3 10 1 0
3 11 1 0
4 12 1 0
4 13 1 0
4 14 1 0
M END
[Interfaceから実行する場合]
n-butane.molファイルをCONFLEX Interfaceを用いて開きます。

Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。次に、詳細設定ダイアログの右下にあるをクリックしてください。

表示されたダイアログに、キーワード「PSEUDO_TORS=(1,2,3,4,150.0,100.0)」を追加します。このキーワードにより、C-C-C-C二面角の中心値を150.0、力の定数を100.0として、二面角に制約を課します。
計算設定が終わりましたら、をクリックします。

[コマンドラインから実行する場合]
計算設定は、n-butane.iniファイルにキーワードを記述することで行います。
n-butane.iniファイル
MMFF94S PSEUDO_TORS=(1,2,3,4,150.0,100.0)
「PSEUDO_TORS=(1,2,3,4,150.0,100.0)」は、C-C-C-C二面角の中心値を150.0、力の定数を100.0として、二面角に制約を課すことを意味します。
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
n-butane.molとn-butane.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par n-butaneenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
力定数の値を大きくするほど、最適化後の構造パラメーターは中心値に近くなります。定数値を変えて最適化した後の二面角を表1に示します。
表1:力の定数と最適化後のC-C-C-C二面角
| 力の定数 (kcal・mol-1・rad-2) | 二面角 (°) |
|---|---|
| 10 | 168.6 |
| 100 | 153.4 |
| 1000 | 150.3 |
*分子単位での構造固定
複数の分子を含む系の構造最適化において、特定の分子のみ構造最適化を行い他の分子は固定する方法を説明します。
キーワードは、最適化のオプションである「OPT=GROUP」と、「MOL_GROUP=(I,N)」を設定する必要があります。ここで、Nは構造最適化を行いたい分子のグループ番号、Iはその分子の原子番号です。
計算例
α-D-グルコースと水を含む系について、水分子のみ構造最適化します。
α-D-グルコースに水分子が配位した3次元構造

座標データ(aDglucose_H2O.mol)
aDglucose_H2O.mol
27 26 0 0 0 999 V2000
-0.5024 2.4148 0.6016 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.8520 -1.0932 -0.5731 O 0 0 0 0 0 0 0 0 0 0 0 0
0.4519 -1.5744 -0.2774 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.2020 1.2411 -0.1553 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.1949 0.1125 0.1452 C 0 0 0 0 0 0 0 0 0 0 0 0
2.1431 1.8092 -0.2200 O 0 0 0 0 0 0 0 0 0 0 0 0
0.5330 -1.9764 1.0847 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8319 -0.9820 -0.1321 O 0 0 0 0 0 0 0 0 0 0 0 0
-2.6186 0.5024 -0.2701 C 0 0 0 0 0 0 0 0 0 0 0 0
1.5391 -0.5233 -0.5592 C 0 0 0 0 0 0 0 0 0 0 0 0
1.2184 0.7799 0.1723 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.5094 -0.5881 -0.0183 O 0 0 0 0 0 0 0 0 0 0 0 0
0.2794 2.9953 0.4978 H 0 0 0 0 0 0 0 0 0 0 0 0
0.6445 -2.4621 -0.8896 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.2535 1.5201 -1.2154 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.2141 -0.1177 1.2184 H 0 0 0 0 0 0 0 0 0 0 0 0
3.0267 1.3889 -0.1951 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.0183 -2.7768 1.1469 H 0 0 0 0 0 0 0 0 0 0 0 0
2.6839 -1.4193 0.7318 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.6702 0.7196 -1.3421 H 0 0 0 0 0 0 0 0 0 0 0 0
-2.9791 1.3731 0.2851 H 0 0 0 0 0 0 0 0 0 0 0 0
1.6027 -0.3262 -1.6358 H 0 0 0 0 0 0 0 0 0 0 0 0
1.3472 0.6668 1.2558 H 0 0 0 0 0 0 0 0 0 0 0 0
-3.0727 -1.3725 -0.4004 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.3106 0.8234 -2.7522 O 0 0 0 0 0 0 0 0 0 0 0 0
-0.3290 0.0085 -2.2451 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.9697 0.7822 -3.4490 H 0 0 0 0 0 0 0 0 0 0 0 0
1 4 1 0 0 0 0
1 13 1 0 0 0 0
2 3 1 0 0 0 0
2 5 1 0 0 0 0
3 7 1 0 0 0 0
3 10 1 0 0 0 0
3 14 1 0 0 0 0
4 5 1 0 0 0 0
4 11 1 0 0 0 0
4 15 1 0 0 0 0
5 9 1 0 0 0 0
5 16 1 0 0 0 0
6 11 1 0 0 0 0
6 17 1 0 0 0 0
7 18 1 0 0 0 0
8 10 1 0 0 0 0
8 19 1 0 0 0 0
9 12 1 0 0 0 0
9 20 1 0 0 0 0
9 21 1 0 0 0 0
10 11 1 0 0 0 0
10 22 1 0 0 0 0
11 23 1 0 0 0 0
12 24 1 0 0 0 0
25 26 1 0 0 0 0
25 27 1 0 0 0 0
M END
[Interfaceから実行する場合]
aDglucose_H2O.molファイルをCONFLEX Interfaceを用いて開きます。

Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。次に、詳細設定ダイアログの右下にあるをクリックしてください。
表示されたダイアログに、キーワード「OPT=GROUP」「MOL_GROUP=(25,1)」「NOSYMMETRY」を追加します。

「OPT=GROUP」「MOL_GROUP=(25,1)」を指定することで、25番原子を含む水分子のみが構造最適化されます。「NOSYMMETRY」は、最適化後の構造について対称性を判定するための座標変換を行わないことを意味します。これにより、最適化を行っていない𝛼-D-グルコースの原子座標は入力データから変化しません。
計算設定が終わりましたら、をクリックします。
[コマンドラインから実行する場合]
計算設定は、aDglucose_H2O.iniファイルにキーワードを記述することで行います。
aDglucose_H2O.iniファイル
MMFF94S OPT=GROUP MOL_GROUP=(25,1) NOSYMMETRY
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。「OPT=GROUP」「MOL_GROUP=(25,1)」を指定することで、25番原子を含む水分子のみが構造最適化されます。「NOSYMMETRY」は、最適化後の構造について対称性を判定するための座標変換を行わないことを意味します。これにより、最適化を行っていないα-D-グルコースの原子座標は入力データから変化しません。
aDglucose_H2O.molとaDglucose_H2O.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par aDglucose_H2Oenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
水分子のみを最適化した構造を下記に示します。可視化の方法は、「計算結果の可視化」をご覧ください。

*原子座標固定による構造固定
ここでは原子座標を固定する方法について説明します。座標固定をする原子は、キーワード「FIXED_ATOMS=(I,J,...)」により指定します。
あるいは、最適化する原子を明示的にキーワード「OPTMZD_ATOMS=(I,J,...)」で指定することで、他の原子を固定することもできます。ここで、IやJは原子の通し番号です。なお、両キーワードは同時に利用できません。両キーワードが指定された場合(iniファイルに記述した場合)、両キーワードによる設定は無効となります。原子の番号を指定する際は、「K-L」のようにハイフンを用いると、K番からL番までの原子について指定できます。
計算例
カプトプリルについて、5員環を固定した構造最適化を行います。
カプトプリルの3次元構造

座標データ(Captopril.mol)
Captopril.mol
29 29 0 0 0 0 0 0 0 0999 V2000
-2.3252 -1.0023 1.2229 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.6239 -0.4662 -0.4492 N 0 0 0 0 0 0 0 0 0 0 0 0
-2.0498 -0.4139 -0.1391 C 0 0 0 0 0 0 0 0 0 0 0 0
-2.6884 -1.2552 -1.2306 C 0 0 0 0 0 0 0 0 0 0 0 0
-1.5887 -2.2017 -1.6972 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.3763 -1.8473 -0.8534 C 0 0 0 0 0 0 0 0 0 0 0 0
-3.4559 -1.0423 1.6461 O 0 0 0 0 0 0 0 0 0 0 0 0
-1.3170 -1.4796 1.9618 O 0 0 0 0 0 0 0 0 0 0 0 0
0.2831 0.5566 -0.3768 C 0 0 0 0 0 0 0 0 0 0 0 0
1.7272 0.3117 -0.7399 C 0 0 0 0 0 0 0 0 0 0 0 0
2.5188 1.6023 -0.5751 C 0 0 0 0 0 0 0 0 0 0 0 0
-0.0725 1.6553 -0.0222 O 0 0 0 0 0 0 0 0 0 0 0 0
2.3057 -0.7585 0.1764 C 0 0 0 0 0 0 0 0 0 0 0 0
2.4156 2.1604 1.1489 S 0 0 0 0 0 0 0 0 0 0 0 0
-2.4462 0.6256 -0.1090 H 0 0 0 0 0 0 0 0 0 0 0 0
-3.0306 -0.6102 -2.0707 H 0 0 0 0 0 0 0 0 0 0 0 0
-3.5838 -1.8050 -0.8634 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.8868 -3.2604 -1.5267 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.3835 -2.0978 -2.7862 H 0 0 0 0 0 0 0 0 0 0 0 0
0.5587 -1.9230 -1.4524 H 0 0 0 0 0 0 0 0 0 0 0 0
-0.2408 -2.5317 0.0138 H 0 0 0 0 0 0 0 0 0 0 0 0
-1.7170 -1.8038 2.7862 H 0 0 0 0 0 0 0 0 0 0 0 0
1.7909 -0.0300 -1.7972 H 0 0 0 0 0 0 0 0 0 0 0 0
3.5838 1.4203 -0.8424 H 0 0 0 0 0 0 0 0 0 0 0 0
2.0958 2.3847 -1.2441 H 0 0 0 0 0 0 0 0 0 0 0 0
3.3709 -0.9399 -0.0905 H 0 0 0 0 0 0 0 0 0 0 0 0
1.7276 -1.7018 0.0555 H 0 0 0 0 0 0 0 0 0 0 0 0
2.2416 -0.4159 1.2334 H 0 0 0 0 0 0 0 0 0 0 0 0
3.1721 3.2604 0.9861 H 0 0 0 0 0 0 0 0 0 0 0 0
3 1 1 0
1 7 2 0
1 8 1 0
2 3 1 0
2 6 1 0
2 9 1 0
3 4 1 0
3 15 1 0
4 5 1 0
4 16 1 0
4 17 1 0
5 6 1 0
5 18 1 0
5 19 1 0
6 20 1 0
6 21 1 0
8 22 1 0
9 10 1 0
9 12 2 0
10 11 1 0
10 13 1 0
10 23 1 0
11 14 1 0
11 24 1 0
11 25 1 0
13 26 1 0
13 27 1 0
13 28 1 0
14 29 1 0
M END
[Interfaceから実行する場合]
Captoril.molファイルをCONFLEX Interfaceを用いて開きます。
Calculationメニューから「CONFLEX」を選択し、開いた計算設定ダイアログのをクリックします。
次に、詳細設定ダイアログの右下にあるをクリックしてください。
表示されたダイアログに、キーワード「FIXED_ATOMS=(2-6)」を追加します。

「FIXED_ATOMS=(2-6)」により、環構造部分を固定した構造最適化を行うことができます。
なお、環構造の固定は、キーワード「FIXED_ATOMS=(2-6)」を追加する以外に、「FIXED_ATOMS=(2,3,4,5,6)」または「OPTMZD_ATOMS=(1,7-29)」としても設定することができます。
計算設定が終わりましたら、をクリックします。
[コマンドラインから実行する場合]
計算設定は、Captoril.iniファイルにキーワードを記述することで行います。
Captoril.iniファイル
MMFF94S FIXED_ATOMS=(2-6)
「MMFF94S」は、MMFF94s力場を用いて計算を行うことを意味します。
「FIXED_ATOMS=(2-6)」により、環構造部分を固定した構造最適化を行うことができます。なお、環構造の固定は、キーワード「FIXED_ATOMS=(2-6)」を追加する以外に、「FIXED_ATOMS=(2,3,4,5,6)」または「OPTMZD_ATOMS=(1,7-29)」としても設定することができます。
Captoril.molとCaptoril.iniの二つのファイルを一つのフォルダに格納し、下記コマンドを実行してください。計算が始まります。
C:\CONFLEX\bin\conflex-10a.exe -par C:\CONFLEX\par Captorilenter
上記は、Windowsの場合です。他OSにおける実行コマンドについては、本書の「実行方法」を参照してください。
計算結果
環部分(No.2-6)以外を最適化した構造とその座標データを下記に示します。可視化の方法は、「計算結果の可視化」をご覧ください。
環部分(No.2-6)以外を最適化した構造

環部分(No.2-6)以外を最適化した構造の座標データ(Captopril-F.mol)
Captopril.mol
NONE,E = 11.183, G = 4.571E-08, M(0) MMFF94S(2010-12-04HG)
29 29 0 0 999 V2000
-2.4430 -0.8871 1.2669 C 0 0 0 0 0
-0.6239 -0.4662 -0.4492 N 0 0 0 0 0
-2.0498 -0.4139 -0.1391 C 0 0 0 0 0
-2.6884 -1.2552 -1.2306 C 0 0 0 0 0
-1.5887 -2.2017 -1.6972 C 0 0 0 0 0
-0.3763 -1.8473 -0.8534 C 0 0 0 0 0
-3.5862 -0.9722 1.6925 O 0 0 0 0 0
-1.4082 -1.2283 2.0621 O 0 0 0 0 0
0.2367 0.5531 -0.1333 C 0 0 0 0 0
1.7337 0.3050 -0.3298 C 0 0 0 0 0
2.5093 1.6332 -0.4281 C 0 0 0 0 0
-0.1962 1.6320 0.2729 O 0 0 0 0 0
2.2822 -0.6428 0.7379 C 0 0 0 0 0
2.9575 2.4405 1.1463 S 0 0 0 0 0
-2.3907 0.6266 -0.2018 H 0 0 0 0 0
-2.9966 -0.6195 -2.0696 H 0 0 0 0 0
-3.5750 -1.8028 -0.8948 H 0 0 0 0 0
-1.8649 -3.2542 -1.5802 H 0 0 0 0 0
-1.3807 -2.0322 -2.7601 H 0 0 0 0 0
0.5549 -1.9446 -1.4157 H 0 0 0 0 0
-0.3095 -2.4758 0.0407 H 0 0 0 0 0
-1.8251 -1.4249 2.9265 H 0 0 0 0 0
1.8469 -0.1748 -1.3104 H 0 0 0 0 0
3.4547 1.4484 -0.9511 H 0 0 0 0 0
1.9539 2.3588 -1.0328 H 0 0 0 0 0
3.3735 -0.7125 0.6778 H 0 0 0 0 0
1.8903 -1.6560 0.6140 H 0 0 0 0 0
2.0042 -0.3222 1.7476 H 0 0 0 0 0
3.3407 3.6094 0.6130 H 0 0 0 0 0
3 1 1 0 0
1 7 2 0 0
1 8 1 0 0
2 3 1 0 0
2 6 1 0 0
2 9 1 0 0
3 4 1 0 0
3 15 1 0 0
4 5 1 0 0
4 16 1 0 0
4 17 1 0 0
5 6 1 0 0
5 18 1 0 0
5 19 1 0 0
6 20 1 0 0
6 21 1 0 0
8 22 1 0 0
9 10 1 0 0
9 12 2 0 0
10 11 1 0 0
10 13 1 0 0
10 23 1 0 0
11 14 1 0 0
11 24 1 0 0
11 25 1 0 0
13 26 1 0 0
13 27 1 0 0
13 28 1 0 0
14 29 1 0 0
M END