Gaussian計算結果の表示
ここでは、「Results」メニューとそれらのオプションに関して説明します。このメニューを用いると、いろいろなダイアログボックスが表示され、 Gaussianの出力ファイルから計算結果を調査することができます。ファイルに必要な項目が含まれていない場合、メニュー項目はグレーで表示されます。
表示されるスペクトルやプロットは、同じ手順で操作できます。これらの操作方法は、このマニュアルの最後のセクションで説明します。
計算の要約表示
メニューから「Results=>Summary」と選択すると、Gaussianの計算結果の要約が表示されます(Gaussianのログとチェックポイントファイルを読み込んだ時のみ)。例を、図73に示します。
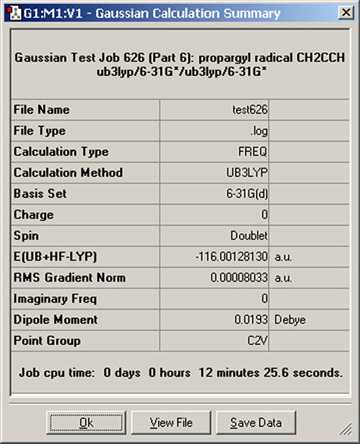
図73 Gaussian計算の要約(Summary)
このウィンドウは、「B3LYP/6-31G(d)」での振動計算の結果を要約した物です。
メニューから「Results=>View File」と選択するか「View File」ボタンを押すと、Gaussian計算のログファイルを開くことができます。
Gaussianで計算された原子の部分電荷の表示
メニューから「 Results=>Charge Distribution」と選択すると、「Display Atomic Charges」ダイアログが開きます(図74)。このツールを用いて、Gaussianの様々な手法で計算された部分電荷密度の表示を設定します。デフォルトはMulliken電荷の表示ですが、他の計算された電荷も表示できます。「Charge Type」メニューに、使用できる選択肢がリストされますので、選択してください。
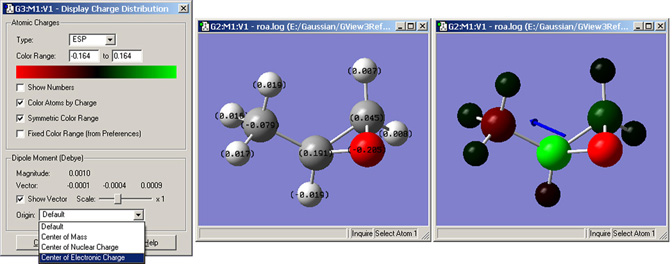
図74:原子の部分電荷の表示
左側のダイアログでは、表示させる電荷の指定とその表示様式を設定できます。中央のウィンドウは、数値での電荷表示です。右のウィンドウは、原子を電荷で色分けしています(設定は、左ダイアログウィンドウに表示されています)。同時に、双極子モーメントベクトルも表示されています。
デフォルトで、電荷表示の色スペクトルは計算された分子の最大電荷を読み込んで電荷に合うように範囲が決められます。この範囲は「Charge Range」フィールドに値を入れることによって、調整可能です。
このダイアログ上部の残ったチェックボックスは、それぞれ以下の意味を持っています:
- Show Charge Numbers:各原子に重ねて部分電荷の数値を表示
- Color Atoms by Charge:「Color Range」フィールドと「Symmetric Color Range」チェックボックスの設定に従い、各原子を電荷によって色分け
- Symmetric Color Range:実際の原子電荷の値を無視して、電荷の範囲を正負同じ絶対値に強制
- Fixed Charge Range:電荷表示を、デフォルトの範囲を使うように強制。デフォルトでは、-1.0から1.0に設定されており、これは「Preferences」の「Charge Distribution」で調整可
ダイアログの「Dipole Moment」エリアでは、双極子モーメントを表すベクトル表示を調整できます。ベクトルが表示されている場合、ベクトルの長さの倍率(デフォルトは1.0)や始点を指定できます。
ダイアログの最下部のボタンで、ダイアログを閉じたあと表示を保持するかどうか制御できます。「Close」ボタンをクリックすれば、原子電荷の表示はそのまま保持され、「Cancel」ボタンをクリックすると元の状態のウィンドウに戻されます。
「Charge Distribution」設定
「Preferences」パネルの「Charge Distribution」で、「Display Atomic Charges」ダイアログの各項目を設定できます(図75)。
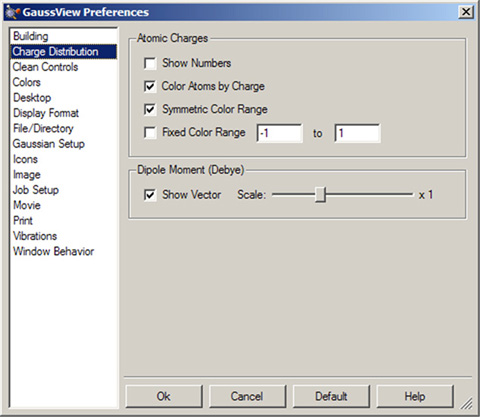
図75:「Charge Distribution」設定
この設定パネルで、「Charge Distribution」ダイアログと同じオプションを使用して、原子電荷の計算結果の表示方法が設定できます。
表面および等高線図の表示
メニューから、「Results=>Surfaces/Contours」と選択すると、「Surfaces and Contours」ダイアログが表示されます(図76)。これを用いて、様々なデータを3次元表示できます。面のデータは、Gaussianのcheckpointファイルを使用して作成するか、cubeファイルを読み込みます。実際に面を表示させるには、次の二つのステップが必要です:
- 「cube」データの作成あるいは読み込み
- 表示用に実際の面を作成
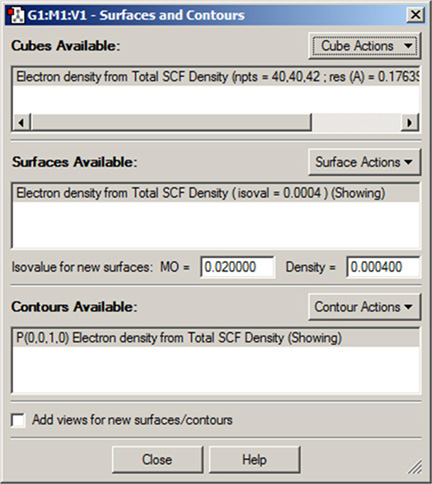
図76「Surfaces and Contours」ダイアログ
このダイアログにより、面や等高線を表示させるためのcubeを選択できます。
また、現在表示されている面と等高線を操作できます。
「Cube」データの作成と操作
以下の「Surfaces and Contours」ダイアログボックスの項目が、「cube」に関連しています:
- Cubes Available:GaussViewで現在表示できるデータが示されます。この項目は、「New Cube 」オプションを使用してcubeデータを作成した時と既に保存されているcubeファイルを読み込んだときに現れます。
- Cube Actions:このボタンにより、cubeの新規作成(New Cube)・外部ファイルからのcubuの読み込み(Load Cube)・cubeデータの外部ファイルへの書き込み(Save Cube)・「Cubes Available」リストからの削除(Remove Cube)が行えます。
「Cube Actions」ボタンの「New Cube」を選択すると、図77のようなダイアログが表示されます。このダイアログで、チェックポイントファイルに含まれる電子密度などのデータから、新規にcubeデータを作成できます。ここでGaussViewは、「cubegen」ユーティリティを自動的に起動します。
「Type」ポップアップメニューでは、作成するcubeデータの分子特性を選択します。ほとんどの選択肢では、追加の選択項目が表示され、希望するデータを指定します。例えば図76に示したように、総電子密度(Total Density)のcubeファイルを作成する際には、密度行列の種類を選択できる事が分かります。同様に、分子軌道の場合は、特定の軌道を指定することができます。
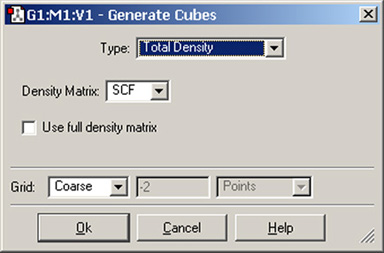
図77「Generate Cubes」ダイアログ
このダイアログの「Type」で指定したcubeファイルが作成されます。
ここでは、「SCF」電子密度で計算した総電子密度のcubeファイルを作成しています。
ここの「Grid」設定では、作成されるcubeファイルの密集度を指定できます。密集度を高くすれば、計算時間も長くなります。通常は、デフォルト設定の「Coarse(粗い)」が、ほとんどの表示目的に適切でしょう。印刷やプレゼンテーションの目的には、「Medium(中間)」が適切でしょう。二番目のフィールドには、「Coarse」「Medium」「Fine(高精細)」それぞれにおける各辺のポイント設定に対応する数値が表示されています。各辺の数値細かく調整したい場合は、「Custom」を選択してください。
「Ok」ボタンをクリックするとダイアログが閉じ、バックグラウンドで計算が開始されてcubeファイルが作成されます。計算が終了したら新しい cubeが「Cubes Available」リストに現れます。ジョブは、メニューから「Calculate=>Current Jobs 」を選択するか対応する「Calculate=>Current Jobs」ボタンを押す事で確認できます。
注:このようにして作成したcubeは、自動ではディスクに保存されません。手動できちんと保存する必要が有ります。そうしないと、GaussViewを終了するときに失われてしまいます。
また「Type」ポップアップメニューには他にもいくつか項目が有り、一つあるいは複数のcubeを変換することができます。既存のcubeを拡大縮小したり二乗したりできます。また複数のcubeの足し合わせや差分を計算できます。例えば、密度の違いを表示させたい場合は、まず気相と溶媒中でそれぞれ計算した密度のような2つのcubeファイルを読み込みます。そして、「Cube Actions=>New Cube」で新しいcubeを作成してから「Subtract Two Cubes」を「Type」メニューから選択します。
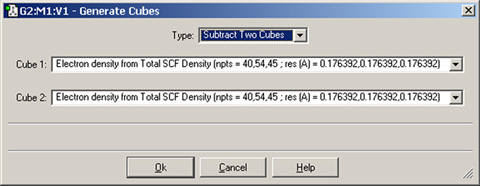
図78 密度差を表すCubeの作成
このダイアログでは、元となる2つのcubeに含まれる値の差から構成される新しいcubeを作成します。
面の表示
以下の「Surfaces and Contours」ダイアログボックスの項目が、面に関連しています:
- Surfaces Available:作成された面のリストを表示
- Isodensity for new surfaces:新しい面を作成する際の、同密度値を指定
- Add views for new surfaces:チェックが入っていると、新しい面を表示する際に新しいウィンドウが開く。チェックされていなければ、現在のビューウィンドウに面が表示される
- Surface Actions:このボタンを押すと、面に関連する機能のメニューが表示される。実際にビューウィンドウ内に面を作成する際に使用。一度作成されたら、面はオリジナルのビューウィンドウに連携される
「Surface Actions」メニューには、以下の項目が含まれます:
- New Surface:選択したcubeを使用して新しい面を作成。ダイアログの関連するフィールドの設定を参照。例えば、同密度値と「add new view」の選択など
- New Mapped Surface:新しく、属性がマッピングされた面を作成(以下参照)
- Show Surface:ビューウィンドウ内で、指定した面を表示
- Hide Surface:ビューウィンドウ内で、指定した面を隠す。しかし、データは保持され、後で再表示可能
- Remove Surface:指定した面を破棄。ビューウィンドウから削除され、「Surfaces Available」リストからも削除
図79に、面の表示例が示されています。
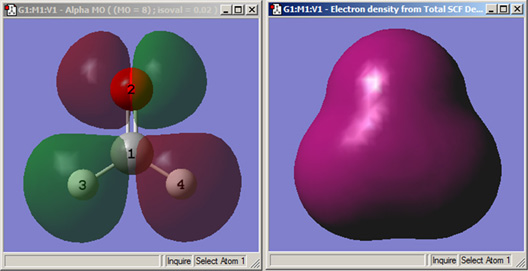
図79 GaussViewで作成した面の例
両方の分子は、ホルムアルデヒドです。
左図は、HOMO分子軌道面を透明モードで表示しています。
右図は、ソリッドモードで電子密度面を表示しています。
面は保存できず、cubeデータのみが保存できます。そのため後で再び表示させたい時は、面の再計算が必要です。
マッピングされた面
GaussViewでは、ある属性値を異なった属性で作成された等値面上にマップできます。これは、「Surface Actions」ボタンのメニューから「New Mapped Surface」を選択する事により実行できます。選択すると、図80のようなダイアログが表示されます。
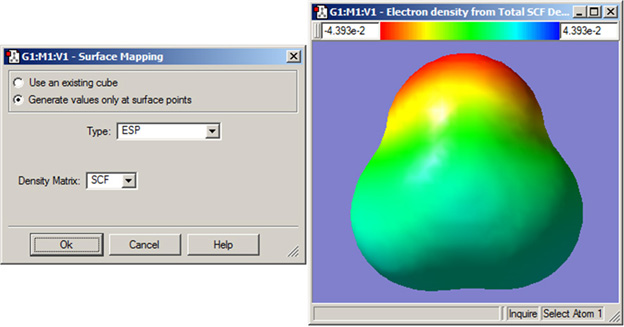
図80 マッピングされた面の作成
このダイアログに表示されている二番目の属性の値で色分けされた面が作成されます。右のウィンドウに、ホルムアルデヒドの静電ポテンシャルの値で色分けされた電子密度面が示されています。
このダイアログの各フィールドは、以下の目的で使用します:
- Use an existing cube:色付けデータとして、存在するcubeから1つを使用。この項目を選択した場合は、リストから希望する面を選択
- Generate values only at surface points:面の各ポイントのみで色づけデータを新たに作成。通常こちらを選択た方が処理が速いですが、Gaussian 03に含まれるcubegenユーティリティが必要。ポップアップメニューから色付けする面のタイプを選択してから、表示される他の属性を指定
図80の右のウィンドウには、色付けされた面と色のマッピングのツールバーが表示されています。面の色付けに使用される色は、スペクトル表示の両端のテキストボックスに示されている最小と最大値の間で均等に割り当てています。これらのボックスの値を変更すると、色の割り振りが変化しマッピングされた面の色も変化します。この色のマッピングツールバーも、他のツールバーと同様に、マウスでクリックしたままウィンドウの周りで移動できます。
面表示のカスタマイズ
面は、ソリッドモデル・透明・ワイヤーメッシュで表示できます。表示は、「Display Format」ダイアログの「Surface」パネルで調整できます。パネルには、メニューのView=>Display Formatやツールバーの「Display Format」ボタンでアクセスできます。面を透明にする設定のダイアログの例が、図81に示されています。
ダイアログの「Z-Clip」スライダーを使用して面の前面を消去し、分子表示の内側を見ることができるようになります。この機能は、3つの表示タイプ全てに有効です。
「IsoValues」ポップアップメニューを使用して、正の値・負の値あるいは両方(デフォルト)の表示を選択できます。「Hide backside」チェックボックスは、面の背面を描画するかどうかをコントロールします。チェックを入れると、透明な面の透明度が増します。表示をメッシュにしてチェックボックスを入れたり外したりしてみて、隠れたり現れたりするのを観察してみてください。
面の属性のデフォルト設定は、「Display Format」設定の「Surface」パネルでも行えます(図81)。
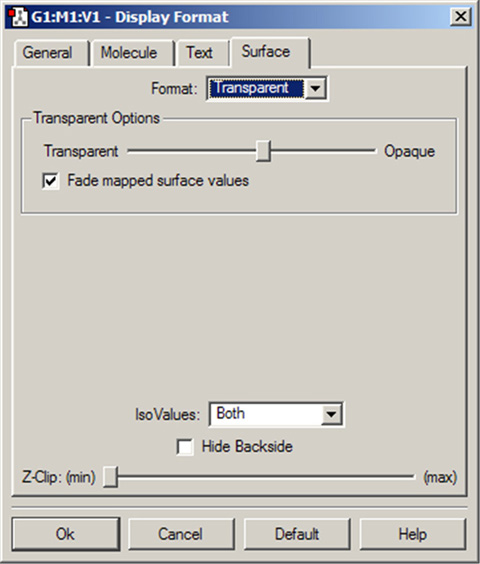
図81 面表示のカスタマイズ
このダイアログで、面の表示方法を調整できます。ここでは、面を透明に表示するよう設定しています。面の透明度は、「Transparent Options」スライドバーで調整できます。「Fade mapped surface values」チェックボックスを入れると、スペクトルの中央値(例えば、ほぼゼロ値)に向けて、透明になっていきます。
等高線図の表示
等高線は、cubeデータの平面への2次元投影です。等高線を表示する際も、「Surfaces and Contours」ダイアログで作成されたcubeデータを使用します(図76)。「Contour Actions」メニューの項目も、「Surface Actions」メニューと類似した動作をします。新しい等高線図を作成するには「New Contour」を選択し、表示させたり隠したりするには「Show Contour」と「Hide Contour」を使用します。等高線を削除するには、「Remove Contour」を選択します。図82に、等高線表示の例を示します。
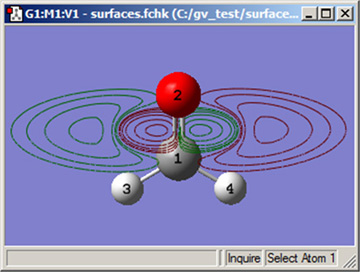
図82 等高線表示の例
この等高線は、HOMOをC=O結合に直行する面に投影しています。
図83に「Contour Actions=>New Contour」を選択した時のダイアログの例を示します。ダイアログには、以下の4つのサブエリアが含まれます:
- 2-D Grid:等高線の各点の値を計算するために用いるグリッドの特性を指定。まず、ポップアップメニューから単位を選択します。そして、グリッドの方向を示す「U」と「V」それぞれの欄に、最小と最大値を指定します。最後に、「Resolution」欄にグリッド間の距離を指定します。
- Plane:等高線を描画する平面を指定します。これに関しては、後で説明します。
- Cube:等高線データの元を、存在しているcubeをリストから選択するのか、平面グリッドに合わせて値を計算するのか、指定します。図83には、前者の例が表示されています。後者は、マッピングした面を作成する際の手順に類似しています(図77参照)。
- IsoValues:等高線の計算する各値を設定します。このリストに数値を追加することもできますし、削除する事も可能です。
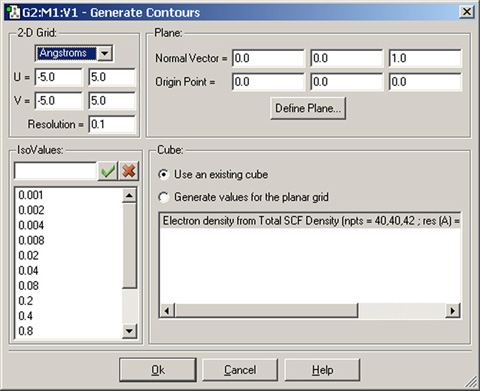
図83 「Generate Contours」ダイアログ
このダイアログでは、作成したcubeや「Surfaces and Contours」ダイアログの「Cube Actions」メニューで読み込んだcubeから、等高線図を作成できます。
等高線を描画する面の指定
デフォルトでは、ビューのXY平面が等高線を描画する面となります。この面は、上のダイアログの「 Plane」エリアで設定できます。面は、ダイアログの「Origin Point」と「Normal Vector」で規定されるベクトルに直行する面として定義されます。デフォルトの「Origin Point」は (0, 0, 0) で、デフォルトの「Normal Vector」は (0, 0, 1) です。
より詳細な面の定義は、「Define Plane」ボタンを押して表示されるダイアログで行えます(図84)。面を定義するには、上で述べた「Normal Vector」で指定する方法と図に示したように面上の3点を入力して指定するという、2通りの方法が有ります。後者の方法では、原点と他の2点を入力して、面を定義します。通常、それぞれを「O」「P」「Q」と呼びます。一番簡単な方法は、3原子をこれらの「Atoms(s)」に割り振るやり方です。ダイアログの「Atom(s)」には複数の原子を入力することができ、その場合はそれらの平均座標が用いられます。また、任意の座標を手入力する事も可能です。
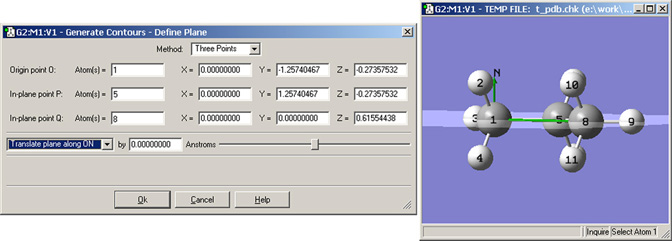
図84 等高線を描画する面の指定
ここの例では、3点を指定する方法で、炭素原子を含む面を定義しています。ビューウィンドウの緑色の線は、基点(O)の法線ベクトル(Nでラベル)そして線OQです。線OPは 原子1と5の間のC-C結合に重なっていて、このビューでは見えていません。
等高線を描画する面が定義できたら、ダイアログ下部のポップアップとコントロールを使用して、いろいろな定義された軸を中心に移動や回転をさせることができます。
振動モードとIR/Ramanスペクトルの表示
メニューの「Results=>Vibrations」を選択すると、振動計算の様々な結果にアクセスできます。図85のような「Display Vibrations」ウィンドウが表示されます。これにより、計算された振動データを動的に表示させることもできます。
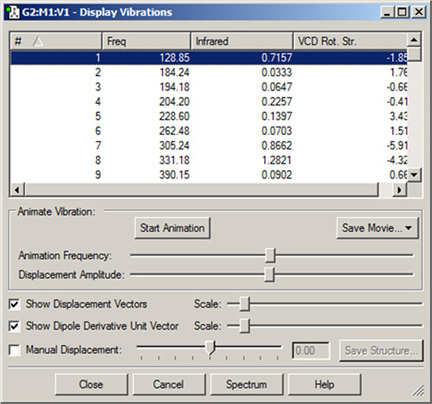
図85 「Display Vibrations」ウィンドウ
このダイアログには、振動計算で得られる全てのスペクトルデータのテーブルが表示されます。
この例では、振動数、赤外強度、ラマン強度、VCD旋光強度が表示されています。ウィンドウの「Spectrum」ボタンを押すと、予測スペクトルがグラフィックで表示されます。
ダイアログには、ディスプレイにどの振動が表示されているのか示されています。振動のアニメーションは、「Start Animation」をクリックすると開始され、「Stop Animation」ボタンをクリックすると停止します。分子の表示は、選択した振動に対応する動きが繰り返されます。「Start/Stop Animation」ボタンの下の2つのスライドバーを使用して、アニメーションのスピードと大きさを設定できます。これらの設定のデフォルトは、「Vibrations」環境設定で調整できます(以下で述べる「Displacement」と「Dipole Derivative Unit」ベクトルの設定も同様)。
また「Save Movie」ボタンを使用すると、アニメーションを動画ファイルやフレーム毎の画像ファイルとして保存できます(詳しくは、このマニュアルで動画の保存について説明した章を参照してください)。
スクロールリストから、他のモードを選択することもできます。その際、アニメーションを止める必要は有りません。また、振動している分子を通常の操作で回転や移動させることもできます。
スペクトルの表示
「Spectrum」ボタンを押すと、Gaussianで計算したスペクトルデータを線画で表示できます。ただし、強度の値は最大値に対する相対値になっていますので、実験で求められたバンド強度に正確な関連付けは行えませんので、注意してください。スペクトル表示の例を、図86に示します。
スペクトルを振動アニメーションさせている場合、目的のピークをクリックすれば、アニメーションの対象を切り替えることができます。
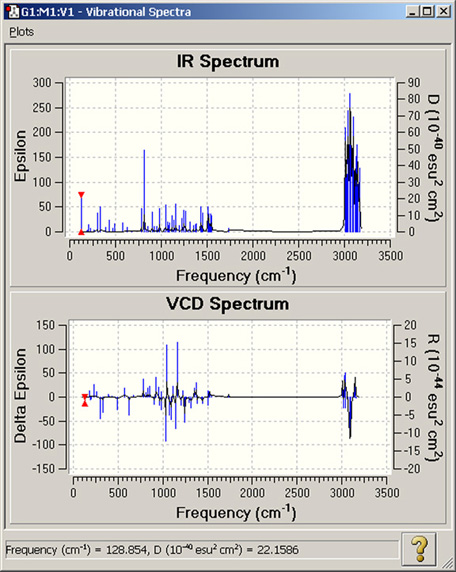
図86 スペクトル表示
これらは、IRとVCDのスペクトルをグラフィック表示しています。表示できるスペクトルは、Gaussianの計算内容に依存します。
様々なベクトル表示
「Display Vibrations」ダイアログの下部に、ベクトル表示関連のものが2つほど有ります(図87参照):
- Show Displacement Vectors:ビューウィンドウに、振動に関連する動きを変位ベクトルで表示
- Show Dipole Derivative Unit Vector:ビューウィンドウに、双極子の微分単位ベクトルを表示
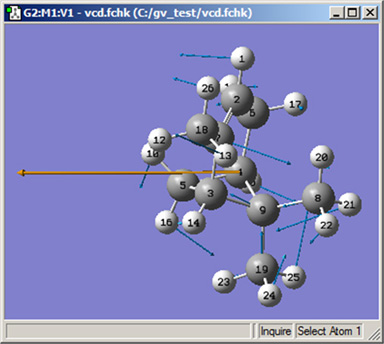
図87 変異ベクトルと双極子の微分単位ベクトルの表示
変異ベクトルは青で、双極子の微分単位ベクトルは左向きのオレンジの矢印で表示されています。
手動での変位表示モード
「Manual Displacement」チェックボックスを入れると、振動の変位表示を手動で設定できるようになります。チェックを入れると、振動のアニメーションは止まります。そして、スライダー・数値ボックス・「Save Structure」ボタンが使用できるようになります。このモードでは、スライダーを動かしたり数値ボックスに値を入力したりすると、選択されている基準振動モードに沿って分子が変形されます。スライダーが中央に有る場合あるいは数値ボックスに0が入力されている状態は、変位していないことを示します。スライダーを右に動かすかあるいは数値欄に正の値を入力すると、現在選択されている基準振動モードの方向に原子が動かされることになります。スライダーを左に動かすかあるいは数値欄に負の値を入力すると、基準振動モードと逆の方向に原子が動かされることになります。ここでの数値は、-1から1の間に制限されています。「Save Structure」をクリックすると、 新規に分子ウィンドウが開き、まさに元のウィンドウに表示されていた変位した構造が表示されます。
このツールの主な用途は、余計な負の固有値を持つ振動を消去する手助けです。遷移状態探索を行う際に、2つ以上の負の固有値を持つ構造になってしまう事がたびたび有ります。これらの誤った負の固有値は通常小さなものですが、真の遷移状態を見つけるには消去しなくてはなりません。そのような場合、負の固有値を持つ誤った振動を選択し、「Manual Displacement」を選択します。スライダーを右に動かすかテキストボックスに正の値を入力し、「Save Structure」をクリックして表示されている構造の新しいファイルを作成し、再計算を行います。この正の方向への原子の移動でうまく行かない場合は、同様の手順で反対の負の方向への移動を試してください。また、移動の大きさも変えてみてください。
手動による変位の2番目の目的は、振動の途中の分子の絵を作成する事です。ある特定の点で、振動アニメーションを止めることができます。
振動に関連する計算の結果
Gaussianの振動計算では、電磁場の摂動において属性をどの振動数で計算するか指定できます。振動数依存の計算結果では「Display Vibrations」ウィンドウの上部にポップアップメニューが追加され、目的とする入射光の振動数を選択できます(図88)。
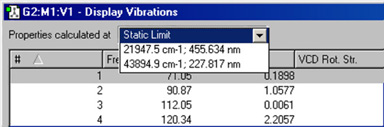
図88 特定の振動数の選択
振動数依存計算の結果を読み込んだ場合、「Display Vibrations」ウィンドウの上部にこのメニューが追加されます。
NMRスペクトルの表示
NMR計算を行った場合、メニューから「Results=>NMR」を選択すると、スペクトルが表示されます(図89参照)。
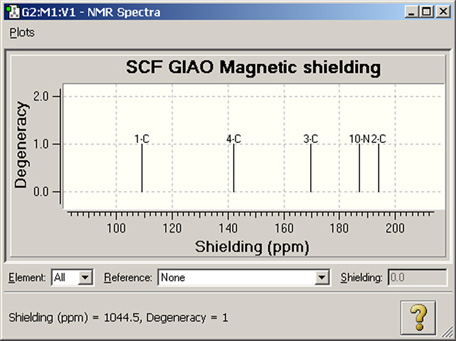
図89 NMRスペクトル表示
これは、デフォルトのNMR表示フォーマットです。計算された全てのピークが表示されています。ピークをクリックすると、ウィンドウの左下に遮蔽値と縮重度が表示されます(上図で表示されているのは2-Cのピークの値)。
NMR Spectraウィンドウの下部の選択欄を使用して、ピークのシフトをTMSや他の基準物質に対する相対値で表示できます(図90)。この機能を使用するには、まず左側のポップアップメニューから目的の元素を選択してください。それから「Reference」で、計算済みの基準物質から一つを選択してください。ピークの位置は、選択した基準物質に対する相対値として反映されます。
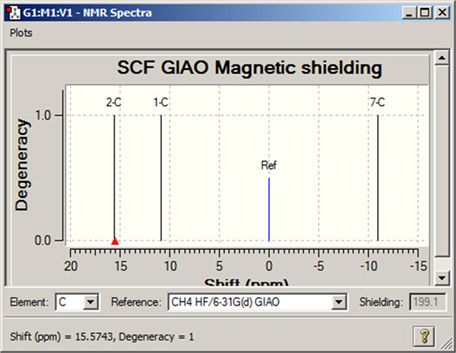
図90 NMR化学シフトの表示
この表示モードでピークをクリックすると、ウィンドウの左下に、選択した基準物質に対する化学シフトの数値が表示されます。対応する遮蔽値は、「Shielding」欄に表示されます。
紫外可視光吸収スペクトルの表示
メニューから「Results=>UV-VIS」を選択すると、励起状態計算で得られるスペクトルが表示されます(図91参照)。
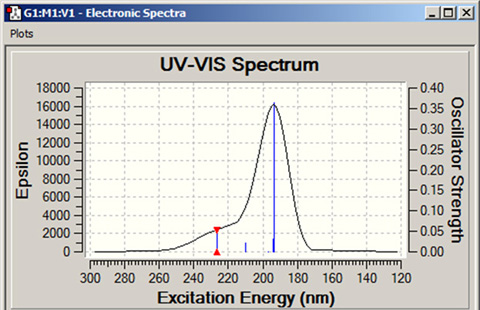
図91 紫外可視光吸収スペクトルの例
構造最適化の結果や連続構造の表示
構造最適化、ポテンシャルエネルギー面のスキャン、IRC・ADMP・BOMDトラジェクトリーなどの計算結果を開く場合、保存されている全ての構造を取り出すには「Open File」ダイアログの「Read intermediate geometries (Gaussian Optimizations Only)」にチェックを入れてください(図92)。その場合、デフォルトでは各構造は新しい分子グループに別々のモデルとして読み込まれます。このチェックボックスは、「Optimizations Only」と表記されていますが、これら全てのジョブタイプに適用されます。
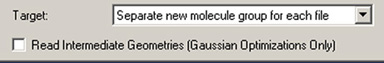
図92 構造最適化および類似ジョブの結果からの中間構造の取り出し
図中の「Target」欄の設定は、複数の構造を含むファイルを開く際の推奨です。ここで「Read Intermediate Geometries」にチェックを入れていないと、最終構造だけが取り出されます。
ビューウィンドウが単一表示モードになっている場合、ツールバーのボタンにより様々な構造をアニメーション表示できます。ツールバーにある緑の円が表示されているボタンをクリックすると、アニメーションが開始します。アニメーションが始まるとボタンが赤い「X」に変わり、それをクリックするとアニメーションが止まります。動画スピードは、「 Display Format」環境設定の「Molecule」パネルにある「Animation Delay」で調整できます。
残りの「Results」メニュー項目(Scan, IRC, Trajectory, Optimization)は、対応する計算により作成された複数の構造のエネルギーや他の項目をプロットしてくれます。図93に、構造最適化のプロットを示します。
表示されるプロットは、ジョブの内容によって変化します:
- 構造最適化:Total Energy(全エネルギー)およびRMS Gradient(エネルギー勾配の平均二乗偏差)
- ポテンシャルエネルギー面スキャン:Scan of Total Energy(スキャン結果の全エネギー値)
- IRC反応経路:Total Energy Along IRC(IRCに沿った、全エネルギー値)とRMS Gradient Along IRC(IRCに沿った、エネルギー勾配の平均二乗偏差)
- トラジェクトリー (ADMPまたはBOMD):Nuclear Kinetic Energy(核運動エネルギー)、Electronic Kinetic Energy(電子運動エネルギー)、Potential Energy(ポテンシャルエネルギー)、Total Energy(全エネルギー)、Total Angular Momentum(全角運動量)、Conservation of Alpha Electrons(アルファ電子の保存性)、Average Alpha Idempotency(アルファ電子の平均べき等元性)、RMS Alpha Idempotency(アルファ電子ベキ等性の平均二乗偏差)、Conservation of Beta Electrons(ベータ電子の保存性)、Average Beta Idempotency(ベータ電子の平均べき等元性)、RMS Beta Idempotency(ベータ電子ベキ等性の平均二乗偏差)。最後の3つは、開核計算の場合のみ。
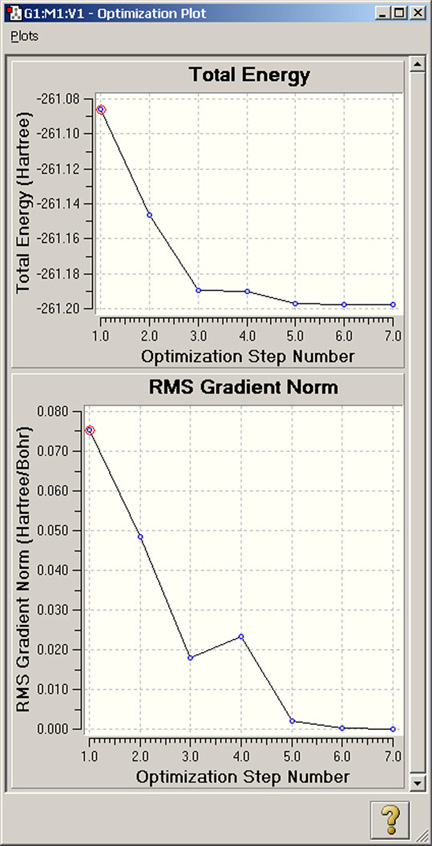
図93 構造最適化からのプロット
プロットには、各最適化ポイントでの、全エネルギーとエネルギー勾配の平均二乗偏差(Root-Mean-Square)が表示されています。
プロット上の点をクリックすると、ビューウィンドウに対応する構造が表示されます。
二変数スキャン計算結果の3次元プロット表示
二変数以上を用いたGaussianのScan計算結果は、GaussViewで3次元の面として表示できます。図94に、例を示します。プロットの各点は、2次元プロットの時と同様に、クリックして選択することができます。
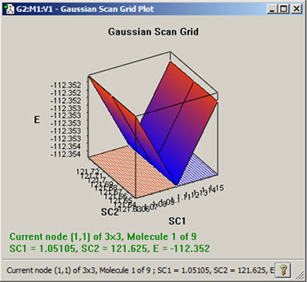
図94 2変数ポテンシャルエネルギー面スキャンのプロット結果
このようなプロットは、複数の変数でのスキャン計算結果のみで可能です。このプロットは、通常のマウス操作で回転や拡大縮小を行うことができます。
プロット項目の追加
上記ジョブタイプでは、様々な属性のプロットを追加できます。それには、プロットウィンドウのメニューから「Plots=>Plot Molecular Property」を選択してください。図95のようなダイアログが表示されます。
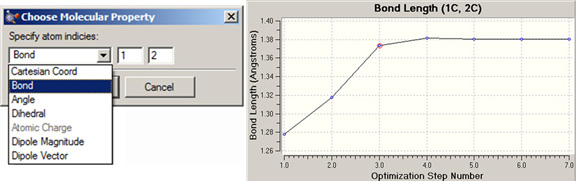
図95 プロット項目の追加
この例は、原子番号1と2のC-C結合距離を構造最適化の進行に対してプロットする設定です。
ダイアログの左側には、プロット可能な様々なアイテムが表示されます。
選択できる項目は、ジョブタイプによって変化します。プロットが作成されたら、プロットウィンドウの標準プロットの下に追加表示されます。
プロットの切れ目の修正
複数の構造を含むファイルを読み込んでプロットした場合、X軸の数値は連続した昇順の値で作成されます。場合によっては、同じ分子グループに複数の別ファイルから分子を読み込んでいる事も有ります。これは、計算の比較のためや前の実行結果に続けて結果を読み込んだ場合などです。その場合、X軸の値は連続した昇順にはならなくなり、プロットもギザギザになってしまいます。この状況がプログラムによって検出されると、メッセージが表示されます。
そのようなプロットを修正するには、メニューの「Plots=>Fix Discontinuity」項目を利用します。この機能を使用した場合、タイトルに「(Modified)」という文字列が加えられプロットに修正が加えられた事を示します。その場合は、注意してプロットを観察してください。
プロットとスペクトルの操作とカスタマイズ
プロットとスペクトルの表示は全て、以下のように様々な方法で操作することができます:
- スクロールとサイズ変更:通常は、同じダイアログに複数のプロット/スペクトルが表示されており、同時に全てを表示できない場合が有ります。その場合、同時により多くのプロットが見られるようにウィンドウのサイズを変更できますし、スクロールバーで下にスクロールさせて、目的のプロットを表示させることもできます。プロット全部のうちいくつかが表示されている状態でウィンドウのサイズを変更すると、X軸が伸縮しプロットの高さはそのまま変わらず、表示領域だけが変更されます。
ダイアログにプロットが一つしか含まれていないか複数含まれているが他が隠されて一つだけが表示されている場合(下図参照)、ウィンドウのサイズ変更を行うとプロットの両軸が伸縮します。特定のプロットを注目したい場合、「Plots」メニューを使用して他のプロットを隠すことができます。
- 特定のプロット/スペクトルを隠す:図96のように、「Plots」メニューの上部には現在表示できる項目が全てリストされています。それぞれの項目名の前に付いているチェックマークを消したり付けたりする事で、特定の項目を隠したり表示させたりできます。メニューの「Show All Plots」を選択すると、全ての項目がチェックされ表示されます。コンテクストメニューの「Hide Other Plots」(下図参照)を選択すると、現在表示されている他のものが全て隠されます。
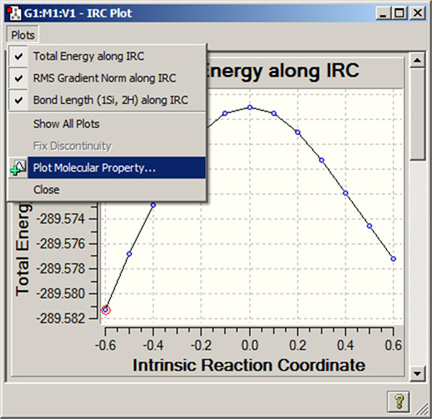
図96「Plots」メニュー
このメニューは、全てのプロットとスペクトルのウィンドウに表示されます。この例は、IRC計算をプロット表示したウィンドウです。この例では、全てのプロットが見えるようになっています。
- 拡大縮小:全てのプロットとスペクトルは、拡大縮小できます。この機能は、細部を観察するのに便利です。拡大するには、左マウスボタンをクリックしたまま拡大したい領域の一角を選択します。マウスボタンを押したまま、拡大したいエリアの対角までドラッグします。拡大エリアは、矩形で囲まれます(図97)。マウスボタンを離すと、選択したエリアがウィンドウ全体へ拡大されます。このプロットは、好きなだけ拡大したままにしておけます。元に戻すには、プロットのコンテクストメニューから「Zoom Out」を選択してください。
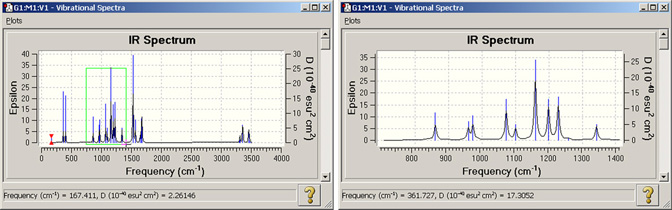
図97 スペクトルの拡大
左側のスペクトルは、GaussViewで最初に表示されたものです。
ここでは、緑のエリアを拡大します。右図がその結果です。
- コンテクストメニュー:プロットやスペクトルの内部やタイトルや軸ラベルが表示されているグレーの部分で右クリックすると、そのプロット/スペクトルに特有のコンテクストメニューが表示されます。メニューのオプションは通常、現在の表示のみに適用されます。コンテクストメニューの例が、図98に示されています。各項目は、以下の機能を持っています:
- Show All Plots:全てのプロットを表示
- Hide Other Plots:現在の項目以外を隠す
- Zoom Out:元のプロット表示に戻し、全ての拡大を取り消し
- Undo Last Zoom:直近の拡大操作を取り消し
- Print:現在のプロットあるいはスペクトルを印刷
- Save Data:プロットあるいはスペクトルデータをテキストファイルに保存。スペクトルの場合、通常のシステムの保存ダイアログにラベル範囲を指定する「Range」ボックスが加えられ、データに含める部分を選択できるようになっています。
- Export:プロットあるいはスペクトルを、グラフィックファイルとして保存
- Properties:プロットの各属性を設定。詳しくは、マニュアルの次章を参照
  con con
図98 プロット/スペクトル表示でのコンテクストメニュー
プロットあるいはスペクトル表示内で右クリックすると、このメニューが表示されます。適用できる項目のみが含まれ使用できる状態になっています。
スペクトルで「Save Data」を選択した場合、右図のような入力欄がファイル保存ダイアログに追加されます。
プロットとスペクトルのカスタマイズ
プロットあるいはスペクトル表示でコンテクストメニューから「Properties」を選択すると、図99のようなウィンドウが表示されます。
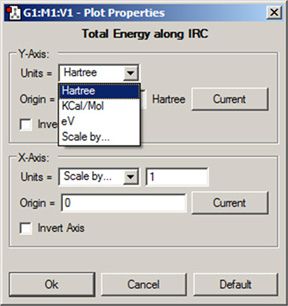
図99 プロット/スペクトルのカスタマイズ
このダイアログを使用して、プロットやスペクトル表示をいろいろカスタマイズできます。ただし全ての項目が常に操作できる訳では有りません。ダイアログの項目は、スペクトルタイプによって変化します。ここでは、振動スペクトルの調整を行っています。
ダイアログの各項目は、以下の機能を持っています:
- Units:対応する軸の単位を選択。ポップアップメニューの「Scale by」を選択してテキストボックスに数値を入力することによって、プロットの座標を拡大縮小できます(図99のX軸参照)
- Origin:対応する軸の始点の値を設定
- 「Current」ボタン:現在選択しているポイントの値を、対応する軸の始点として使用
- Invert Axis:プロットを垂直(Y軸)あるいは水平(X軸)反転
- 「Default」ボタン:全ての項目をデフォルト設定に戻す
ダイアログに表示される場合のある他の項目は、以下になります:
- IR Peak Half-Width at Half Height:振動スペクトルのピーク幅を設定。値を大きくすると、ピークが幅広くなります。
- UV-Vis Peak Half-Width at Half Height:紫外可視光スペクトルのピーク幅を設定。値を大きくすると、ピークが幅広くなります。
- NMR Degeneracy Tolerance:「NMR Spectra」ダイアログのスペクトルピークをグループ化するしきい値を設定。値を大きくすると、より多くのピークが縮重しているとしてグループ化されます。値をゼロにすると、ピークが縮重しているという扱いをしなくなります。
|
