高度な分子構築機能
ここでは、特定の操作や状況のために設計された分子構築機能を説明します。
カスタムフラグメントライブラリーの利用
「Custom Fragment」ボタンにより、手元のフラグメントライブラリーの読み込みと修正を行えます。ライブラリーがアクティブでないときにクリックすると、最後に使用したカスタムフラグメントライブラリーがアクティブになります。このライブラリー内にある最初のフラグメントが現在選択されているフラグメントになり、ツールバーの下の領域と様々な構築ボタンの右側にあるポップアップメニューに表示されます。カスタムフラグメントパレットが開いている時に同じボタンをもう一度クリックすると、図24にあるウィンドウが表示されます。
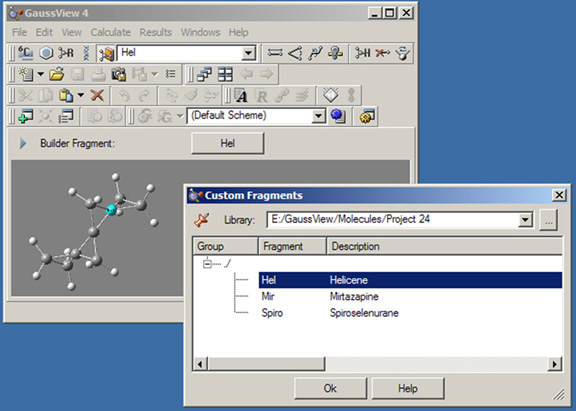
図24.カスタムフラグメントウィンドウ
このパレットにより、開いているライブラリにある利用可能なフラグメントを選択することが出来ます。選択したフラグメントは、ダイアログを閉じるとカレントフラグメントになります。
パレットの上部にある「Library」欄は、利用しているフラグメントライブラリーを示しています。過去に開いたライブラリーもリスト内に表示されます。また、「...」と記されたボタンをクリックすると、追加するライブラリの場所を開くことが出来ます。
フラグメントライブラリーはディスク上では単にフォルダーであり、ライブラリー内の個々のフラグメントはフォルダー内のファイルとして保存されています。つまり、新しいフラグメントライブラリーを作るということは、希望する場所にフォルダーを作ることと、それをパレット内で選択することで構成されます。ライブラリーは、最初は空です。またフォルダー内に有る、フラグメントではないファイルは全て無視されますが、フォルダーはフラグメントファイルの使用に限定した方がいいでしょう。
フラグメントライブラリーにあるフラグメントは、ユーザーが作成した一連のグループにまとめられます。この機能を利用しない場合、全てのフラグメントは”ルート”グループ、すなわち図の「/」内に置かれます。
パレット中央のフラグメントリスト領域内の白い部分を右クリックすることで現れるコンテキストメニューは、フラグメントライブラリーの管理や修正に用いられます。メニューには以下の選択肢があります:
- New Group:選択されているライブラリー内に、新規グループを作成します。グループには一般名が付けられていますが、修正することが出来ます。これと同じ操作をControl-Gで実行できます(Mac OS XではCommand-G)。
- New Fragment From Active Molecule:選択されているグループにある分子を、選択されているフラグメントライブラリーに追加します。一般名欄と説明は、必要に応じて編集することが出来ます。このメニュー項目と同じ機能をControl-N/Command-Nで、実行することが出来ます。
- Edit Cell:選択されている項目の内容(グループ名、フラグメント名、フラグメントの説明)を編集します。最上位のグループ名は修正できませんので、ご注意ください。
- Delete:選択されているフラグメントもしくはフラグメントグループを削除します。最上位のフラグメントグループは削除できませんので、ご注意ください。このメニュー項目と同じ操作をDeleteキーで、行うことが出来ます
ライブラリー内のフラグメントは、一度消去してから再度追加することでしか更新することが出来ませんので、注意してください(すでにあるフラグメントを修正する方法はありません)。
原子リストエディター
原子リストエディターウィンドウは、EditメニューからAtom List項目を選択するか「Atom List」ボタンをクリックすることで表示されます。このツールは、アクティブビューウィンドウにある分子の座標や他のパラメーターを、編集可能なスプレッドシートの形式で表示します。図25に、このウィンドウのデフォルトの構成を示します。
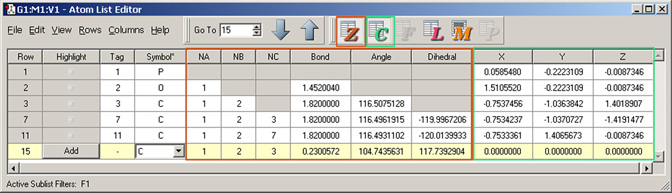
図25:原子リストエディター
表中に表示される列は、ツールバー内のボタンにより調整できます。強調表示されたボタンに対応する列が表示されます。ただし、選択されている分子構造に関係するボタンだけがアクティブになります。個々の列の表示/非表示は、Columnsメニューの項目を用いて設定します。ツールバーの「Go To」コントロールを用いて、特定の原子を直接番号で指定することも出来ます。
このウィンドウのデフォルト構成では、分子のZ-マトリックス座標およびデカルト座標が表示されます。他のデータは、ツールバーボタンを用いて表示するかどうか選択することが出来ます。ボタンは左からそれぞれ、Z-マトリックス座標(Z)、デカルト座標(C)PBC計算用の分率座標(F)、ONIOM層の割り当てとそれに関連した設定(L)、分子力学計算用の原子タイプおよびその他のデータ(M)、そしてPDBファイルの残基と鎖のデータ(P)の表示を制御します。最後のボタンに関連する列は編集できません。図26に、このウィンドウに表示される追加の列を示します。
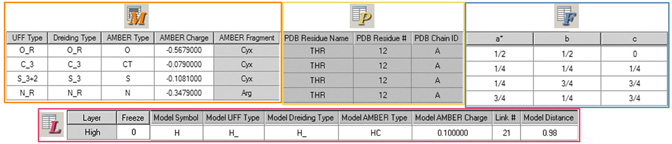
図26.追加される原子リストエディターの列
左上から時計回りに、「Molecular Mechanics Parameters」ボタン、「PDB Info」ボタン、「Fractional Coordinates」ボタン、そして「ONIOM Layers」ボタンによりそれぞれ制御されます。影付きの列は読み込み専用です。
あらゆる分子構造パラメーターは、このスプレッドシート様のウィンドウで編集することが出来ます。左端の列のRowセルをクリックすることで、原子を選択できます。「Highlight」列のドットをクリックすると、対応する原子を分子のビューウィンドウで選択/非選択状態にできます。
ウィンドウの最下部までスクロールし、Symbol列で元素を選択して、Addボタンをクリックすることで、原子を追加することが出来ます。これにより、新しい行がリストに挿入されます(上記図25のように、デフォルトの値が入ります)。
原子リストエディターウィンドウにある各種メニュー項目を以下に記します。
Fileメニューには、表のデータの保存および出力に関連する項目が含まれています:
- Export Data:データをタブで区切られた書式でテキストファイルに保存します
- Print:原子リストの表を出力します
- Close:原子リストエディターのダイアログを閉じます
Editメニュー項目では、分子定義の内容を修正します:
- Undo:一つ前の操作を取り消す
- Redo:一つ前の「取り消した」操作を復元する
- Copy Data:選択されている行のデータをクリップボードにコピーします
- Delete:選択したサブメニュー項目を元に原子を削除します:Selected Atoms(選択した全ての原子)、Selected Hydrogen Atoms(選択した全ての水素原子)、Selected Redundant Atoms(選択した中での複製行)、All Atoms(全原子を削除)、All Hydrogen Atoms(全ての水素原子)、All Redundant Atoms(分子定義内の全ての複製行を消去)
- Reorder:サブメニューで選択した項目にしたがって、表にあるデータの表示順を修正します:
- Selected Atoms by Bonding:選択した原子を、結合している原子が連続して表示されるように並べかえます
- Selected Atoms by Rows:選択した原子を、原子の番号(すなわちTag列の数字)順に並べ替えます
- Selected Atoms: Hydrogens Last:選択した原子の各結合グループ内にある水素原子が下位になるように置き換えます
- All Atoms (Except the First) by Bonding:最初の原子を除いた全原子を、結合している原子が連続して表示されるように並べかえます。
- Atoms by Row:表全体を、原子の番号(すなわちTag列の数字)順に並べ替えます
- All Atoms: Hydrogens Last:各結合グループ内で、全ての水素原子が下位になるように置き換えます
- Z-Matrix:Z-マトリックス分子定義を編集する上で有用な項目が含まれています:
- Standardize:Z-マトリックスを標準的な使用法に準拠した形式に修正します
- Reconnect Selected Atoms by Bonding:結合情報を解析することにより、選択した原子に対するZ-マトリックスのデータを再生成します
- Reconnect Selected Atoms with a Nearly Invalid Dihedral:結合情報を解析することにより、選択した原子の中で2面角があまり妥当でない原子に対するZ-マトリックスを再生成します(Gaussianジョブでまれに必要になります)
- Reconnect All Atoms by Bonding:結合情報を解析することにより、全ての原子に対するZ-マトリックスを再生成します
- Reconnect All Atoms with a Nearly Invalid Dihedral:結合情報を解析することにより、2面角があまり妥当でない全ての原子に対するZ-マトリックスを再生成します(Gaussianジョブでまれに必要になります)
- PBC=>Delete All Atoms Outside Cell:セル内にない原子を削除します。
- PBC=>Translate All Atoms Inside Cell:セル境界線の外にある全ての原子をセル内の等価な位置に移動し、冗長な原子を削除します
- Add Hydrogens for All Atoms:分子構造に水素原子を付加します(主にPDBファイルで使用します)
- Update MM Types for All Atoms:分子力学用原子タイプを再生成します。
「Atom List Editor」を使用している間、「View」メニューによりビューウィンドウ内の原子の強調表示を制御できます。各項目は切り替え式で、メニュー内で項目の隣にチェックマークがある場合はその機能が有効であることを示しています。
- Exclusive Highlighting:テーブル内の一行のみ強調表示を可能とします。Highlight列内の点をクリックすると、それまでの強調表示が消えます
- Z-Matrix Connectivity Highlighting:強調表示された原子それぞれで、その原子のZ-マトリックスの項目(結合、結合角、2面角の定義)を決めるために用いている他の原子も強調表示します。
- Oniom Link Highlighting:行のONIOMレイヤーデータの内容をもとにして、強調表示された原子がONIOM計算においてリンク原子として機能している場合には、ビューウィンドウで[Link]とラベルして表示します
「Rows」メニューは、分子内の複数あるいは全ての原子のテーブル内での表示を制御します。また原子のグループを選択するためのショートカットも提供します:
- Sublist Filters:原子フィルター表記を定義するための「Sublist Filters」ダイアログ(後述します)を開きます。
- Sort Selected:強調表示した原子リストの行を、サブメニューで選択した基準(Ascending by Highlight、またはDescending by Highlight)に準じて並べ替えます。ツールバーにある上向き矢印ボタンと下向き矢印ボタンは、これらのサブメニュー項目に対応します
- Sublist:選択されているサブメニュー項目(Show Sublist AtomsまたはShow Non-Sublist Atoms)により、アクティブなサブリストにある原子を表示するかあるいはそれ以外の原子を表示するかを切り替えます。どちらが選択されているかは、サブメニュー上の対応する項目の隣にあるチェックマークにより示されます
- Select All:全ての原子を無条件に選択します
- Invert Selection:選択されていた全ての原子の選択状態を解除し、選択されていなかった全ての原子を選択状態にします
「Columns」メニューでは、Z-Matrix、Fractional Coordinates、Oniom Data、MM Types、PDB Data各メニュー項目のサブメニューにより、それぞれZ-matrix Coordinatesボタン、Fractional Coordinatesボタン、ONIOM Layersボタン、PDB Infoボタン、Molecular Mechanics Parametersボタンによりテーブルに表示される列をより詳細に設定できます。「Cartesian Coordinates」項目は、対応するボタンと同じ機能を持ちます(個々の座標を分けて選択することは出来ません)。「Help」メニュー項目は、原子リストエディターのヘルプを提供します。
サブリストフィルターの利用
サブリストフィルターは、大きな分子を扱う際に、原子リストエディターで表示する行を扱いやすい数に制限する1つの方法です。原子リストエディターのRows => Sublist Filtersメニューを選択することで表示されるダイアログを用いて、これらのフィルターを定義できます(図27)。
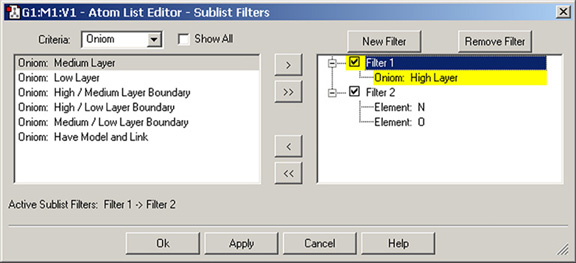
図27.サブリストフィルターの作成
このダイアログにより、原子リストエディター内で表示される行を制限するために用いられるサブリストフィルターの作成および適用が出来ます。この場合、2つのフィルターを連続して適用しています:Filter 1ではHigh ONIOM layerの原子を選択し、Filter 2ではサブセット内の酸素および窒素原子を選択しています。
この機能を使うためには、まずNew Filterボタンを用いてフィルターを作成する必要があります。フィルターは常にFilter nと名付けられ、変更することは出来ません。フィルターは、Criteriaリストにある項目を用いて設定します;ポップアップメニューに、利用可能な基準項目のカテゴリーが表示されます。図では、Filter 1を設定するためにOniomカテゴリーを用いています。リストから一つ以上の基準を選択し、そしてそれらを「 >」ボタンを用いて選択されているフィルターに移動します。ここでは例として示しませんが、異なるCriteriaカテゴリーに属する項目を同じフィルターで組み合わせることが可能です。
「>>」ボタンは、同様にリスト内の全ての項目を移動します。フィルター内の項目は、「 < 」および「<<」ボタンにより、それぞれ選択した項目の削除および全項目の削除が可能です。フィルター全体を削除するには、Remove Filterボタンを用います。
フィルター内の基準は、論理和ORにより組み合わせられます:あらゆる基準にマッチした原子(あるいはそれ以外、原子リストエディターのRows=>Sublistメニューの設定による)を表示します。フィルターは、それぞれの名前の左にあるチェックボックスの選択/非選択によりアクティブまたは非アクティブになります。フィルターが1つのみアクティブな時は、そのフィルターにマッチした全ての原子に作用します。複数のフィルターがアクティブな時は、全てのアクティブなフィルターが、論理積ANDによりダイアログ内で表示されている順に適用されます。例えば、Filter 2はFilter 1を適用した後に残った原子に対してのみ適用されます。後に続くフィルターは、それ以前のフィルターにより生じた選択を拡大せず、さらにふるい分けるのみとなります。
使用できるCriteriaカテゴリーは以下の通りです:
- Tags:一つ以上の原子タグ番号(シリアル番号)を選択します
- Tag Range:連続した範囲の原子タグを選択します
- Tag Select:選択したタグに対応したフィルターを生成します
- Elements:元素タイプにより原子を選択します
- Oniom:ONIOMレイヤー、レイヤーの境界、またはリンクアトムの原子を選択します
フィルターは個々の分子グループに特有で、またフィルターの設定は対応する入力ファイルの異なるセッションには適用されません。言い換えると、入力ファイルを閉じるかまたは再度開いた場合、もしくはGaussViewを終了または再起動した場合は、現在のフィルターの設定は失われます。
結合エディター
結合エディターは、Edit=>Connectionメニューを選択することで表示されます。これは、分子グループ全体で適切な原子の順序付けを維持しているかどうかを調べるために使用することが出来ます。分子グループにわたって関連する構造同士の原子タグをそろえるために、自動的に原子を並べ替えることにも利用できます(QST2やQST3といった遷移状態最適化のための構造を用意するのに便利です)。手動での原子の並べ替えも可能です。
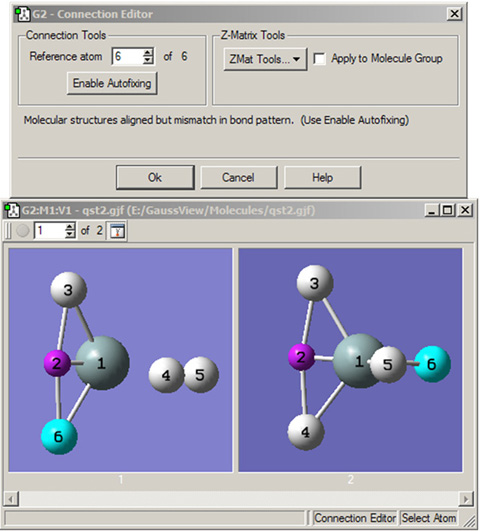
図28.結合エディター
結合エディターは、2つの分子構造で等価な原子を確認や設定することに用います。分子グループを操作するときは、マルチ構造表示モードで見ることにより比較や編集が簡単になります。この不自然な例では、原子を適切な対応関係にするためには片方のモデルの原子4と6の順序を交換する必要があります。この変更は、「Enable Autofixing」ボタンをクリックして行うか、あるいは手動で行うことが出来ます。
結合エディターがアクティブなときは、分子グループ全体で分子の原子タグが一致しているかどうか解析され、ステータスバーに結果が表示されます。修正できそうなミスマッチが検出された場合、ダイアログにこのことが表示されかつユーザーがEnable Autofixingを使用できることが示されます。このボタンをクリックすることで、結合エディターはこれらミスマッチを取り除くために原子ラベルを並べ替えます。自動修正の成功もしくは失敗は、ステータスバーに表示されます。自動修正がアクティブなときは、手動での原子タグの並び替えではいつも自動修正が呼び出され、導入されたあらゆるミスマッチを取り除きます。
自動修正をアクティブにすると、ボタンのラベルがDisable Autofixingに変わり、このボタンをクリックすることで、自動修正を無効にすることが出来ます。自動修正が機能しない場合には、ボタンのラベルはNo Autofixingとなります。
マウスを使って手動で原子を並べ替えることも出来ます。以下の操作は、参照原子を基準として使用します(以下の最初の項目をご参照ください):
- 左クリック:クリックした原子を参照原子に指定します。この操作はReference Atomコントロールを使って行うことも出来ます。
- 中央クリック:クリックした原子のタグを、参照原子より1つ大きな数値に設定します。
- 右クリック:選択した原子のタグと参照原子のタグを交換します。クリックした原子が新たに参照原子となります。
「Apply to Molecule Groupチェックボックスは、結合エディターの操作を現在選択されているモデルのみに適用するかあるいは分子グループ内の全てのモデルに適用するかを決定します。
「ZMat Tools」ポップアップにより、分子あるいは分子グループに対してZ-matrix操作を実行することが出来ます:
- Sort H:水素原子を原子並びの最後に移動します。
- Reorder:標準的なZ-matrix並びに再配列します。
- Reconnect:結合データを再生成します。
このポップアップメニューの他の項目は、Gaussianには適用されません。
冗長座標エディター
冗長座標エディターにより、Gaussianによる最適化あるいはポテンシャルエネルギー曲面のスキャン計算に用いられる冗長座標を作成または編集することが出来ます。Edit=>Redundant Coordinates メニューあるいは「Redundant Coordinate Editor 」ボタンによりアクセスします(図29)。
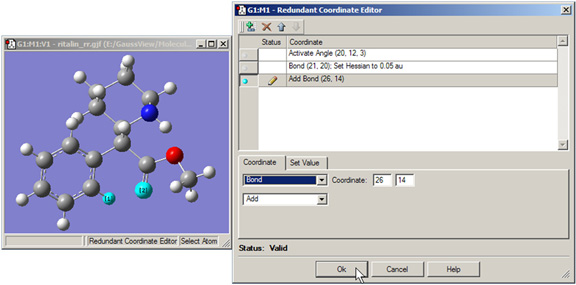
図29.冗長座標エディター
最適化の出力に距離が記録されるように、2つの選択した原子の結合座標を追加しています(原子間で実際に結合してはいません)。ウィンドウ内のリストには、すでに追加してある他の2つの座標が表示されています。
このダイアログの中心的な特徴は、スクロール可能なリスト内に各座標の情報が含まれていることです。「Status」列の鉛筆アイコンは、現在アクティブな座標であることを示す印です。「status」列の警告記号は、座標が不完全かもしくは不適切であることを示しています。現在アクティブな座標が不完全あるいは不適切である場合、底部にあるステータスバーに警告の内容が表示されます。「coordinate」列には、座標やパラメーターの簡単な説明が含まれています。
一番上のツールバーには、新規座標を追加するボタン、座標を削除するボタン、そして項目をリスト内で上あるいは下に移動するボタンがあります。新規座標の追加により未定義の座標が生成し、この座標は現在アクティブな座標のすぐ下に配置されて、アクティブな座標になります。
使用できる全ての冗長内部座標の形式は、Coordinate」パネルにある2つのポップアップ内のさまざまな選択肢によりサポートされています。またこれらのメニューにある項目の意味は一目瞭然です。座標タイプの設定に必要な追加の欄が、座標タイプを選択することで現れます。「Set Value」パネルにより、座標値の指定もしくは現在設定されている値を増やすことが出来ます。
冗長座標エディターを閉じる時に、ある座標が不適切あるいは不完全である場合、そのことが通知されかつ編集を継続するよう促すダイアログが現れます。それでもエディターを閉じると、不適切な座標は削除されます。
ONIOMレイヤーの設定
メニューから「Edit=>Select Layer」項目を選択するかSelect Layer」ボタンをクリックすることで、レイヤー選択ツールが表示されます。このウィンドウは、各ONIOMレイヤーに原子をグラフィカルに割り振ることに用います(図30)。
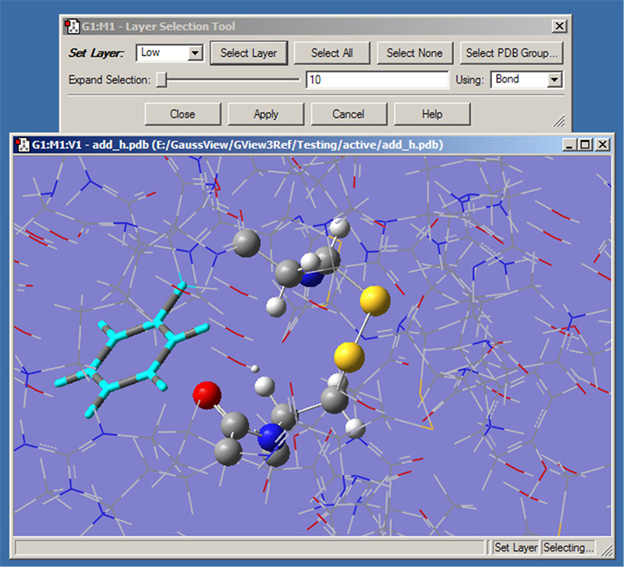
図30.ONIOMレイヤーへの原子の割り振り
このダイアログでは、ONIOM計算で用いる各レイヤーへの原子の割り振りを行うことが出来ます。レイヤーの違いは、表示形式の違いで示されています。ここでは、左側にある環状原子がレイヤーへの割り振りを行う原子として選択されています。
選択した原子を各レイヤーへ割り振るには、いくつかの方法があります:
- ダイアログの一番上にあるボタンを用いて選択を行うことが出来ます。「Select Allボタンは分子内の全原子を選択し、また「Select None」は全原子を非選択状態にします。「Select Layer」ボタンは、設定したレイヤーに割り振られている全ての原子を選択します。「Select PDB Group」ボタンをクリックすると、別の選択用ダイアログが開き、選択されているモデルに対応するPDBファイルに残基の情報がある場合、原子を残基ごとに選択することを可能にします(図31)。
- 原子を手動で選択ないし非選択にするには、マウスの左ボタンを使って、選択したい原子を全て選択するまでクリックします。選択状態の原子を追加するには、Shiftキーを押しながらクリックしてください。
- 近接した原子の選択:原子を1つ選択してからExpand Selectionスライダーを動かすと選択範囲を拡げられます。必要な原子全てを選択するまでスライダーを動かしてください。右に動かすことで、最初に選択した原子からの選択距離が増加します。
距離に基づいた原子の選択では、選択される原子との距離は、ポップアップメニューを用いて選択した以下の基準によって決まります:
- Bond:結合している原子のみを選択します
- Distance:結合しているかどうかにかかわらず、選択している原子に対しての距離のみに基づいて選択します。
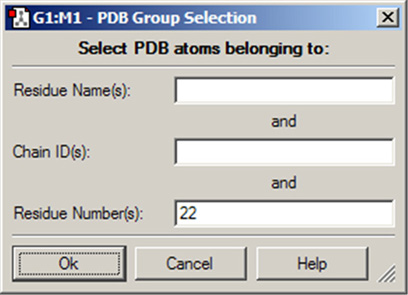
図31 PDB残基情報に基づいた各レイヤーに割り振る原子の選択
このダイアログでは、指定したい残基を残基名、残基番号、および鎖(AまたはB)で指定することが出来ます。省略された項目はワイルドカードとして動作します。例えば、図のように残基番号22のみを入力した場合、両方の鎖の22番残基が選択されます。同様に、残基名のみの入力では分子内にあるその残基が全て選択されます。
レイヤーへの割り振りの確認
一旦原子を選択し、選択したレイヤーに原子を割り振るためには「Apply」ボタンをクリックする必要があります。Apply」をクリックすると、選択状態は解除されることに注意してください。変更した内容を適用せずにパネルを閉じると、それらの内容は消去されます。
レイヤーの表示形式
デフォルトでは、異なるレイヤーの原子は異なる形式でビューウィンドウ内に表示されます。標準設定を用いると、Highレイヤーの原子はボール&スティック形式、Mediumレイヤーの原子はチューブ形式、Lowレイヤーの原子はワイヤーフレーム形式で表示されます。これらの表示形式は、現在選択されている分子でView=>Display Formatを選択するかあるいは、将来表示する全てのビューのために、環境設定にあるDisplay FormatのMoleculeパネル内の対応する欄を設定することで、カスタマイズすることが出来ます。
分子軌道エディター
メニューからEdit=>MOs項目を選択するか「MO Editor」ボタンにより、図32に示したMOsウィンドウが開きます。このウィンドウは、Gaussian計算により得られた分子軌道(MOエネルギーと占有数の図、可視化したMO等値面)などを調べたり、新しいMOを計算することやCASSCF計算に用いる活性空間MOを視覚的に選択することに用いられます。
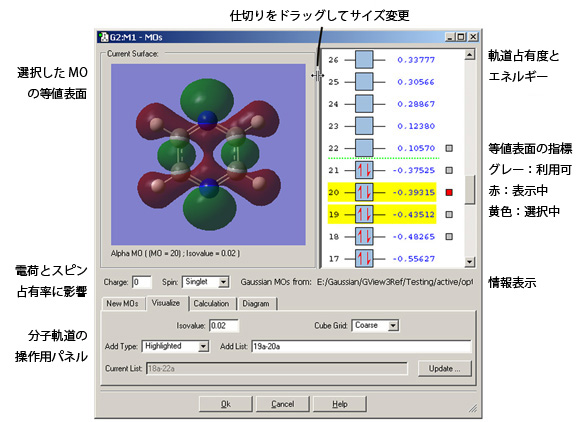
図32 分子軌道エディター
このウィンドウは、分子軌道を視覚的に調べることと、次に行うGaussian計算のための分子軌道の占有数または順序の変更に用いられます。この図では、表面を表示することが可能な5つのMO(かつ選択されている2つのMO)のうちの1つである20番目のMOを表示しています。
「MOs」ダイアログには、分子と(表示可能であれば)選択しているMOに対応した等値面を表示するためのビューウィンドウが含まれています。このビューは読み込み専用で、分子の編集に用いることは出来ません。機能性で見ると標準のビューウィンドウとほぼ同じ機能が含まれており、分子の並進、回転、拡大・縮小や、コンテキストメニューへのアクセスなどが可能です(詳細は、この章の前にあるビューの操作についての議論および「Gaussian計算結果の表示」の章の等値面に関する議論をご覧ください。)
「Current Surface」ビューの大きさは、「MOs」ウィンドウ全体のサイズを調整することで変更することが出来ます。このビューの幅は、ウィンドウとMO順位図との仕切りをドラッグすることで調整することも出来ます。この操作では、2つのウィンドウの相対的な幅が変化します。
マウスの左クリックにより、可視化するMO、将来行う計算へ含めるMO、あるいは他の操作を行うMOを選択することができます。選択されたMOは、黄色で強調表示されます。面を表示することが可能なMOでは、そのMOの右側に小さい四角形が表示されます。その四角形が赤色のMOは、現在表示されているMOです。他のMOの灰色の四角形をクリックすると、そのMOの表面を表示するように「Current Surface」ビューが更新されます。
ウィンドウの下の部分には、以下の4つのパネルがあります:
- New MOs:分子軌道の読み込みもしくは作成に用いられます
- Visualize:目的のMOの表面を作成することが出来ます
- Calculation:今後行うGaussianジョブのためのMOの順序変更をすることが出来ます
- Diagramビュー領域の右側に表示されているエネルギーレベルと軌道占有数を制御します。
図32に示された「Visualize」パネルにおいて、2つの表面作成パラメーター(等密度値およびグリッド密度、それぞれ「Isovalue」および「Cube Grid 」欄で指定)などのいくつかの欄を用いてどの軌道をどのように可視化するかを指定します。「Add Type」ポップアップには、可視化するMOを特定する項目が含まれています。このポップアップにある選択肢は、「Current List」(「Current List」欄にある軌道を再作成する)と「Other(編集可能な「Add List」欄に表示されている任意の軌道の一覧を指定する)を除くと、メニューに書かれているような選択になります。ここでGaussViewは、「Add List」欄と「Add Type」欄の内容が一致するように自動修正します。「Update」ボタンをクリックすると、指定した軌道の表面が作成されます。
図33に、分子軌道エディターにある他のパネルを(一つずつ積み重ねて)示します。それぞれのパネルについては、この節の残りの中で議論します。
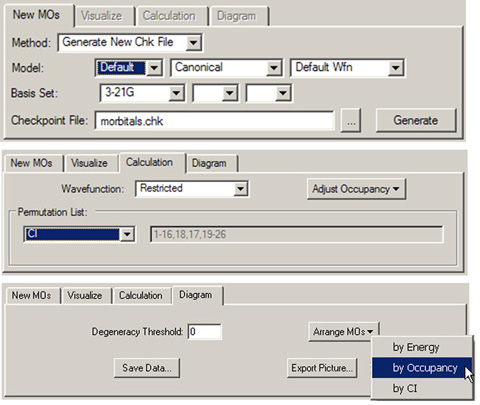
図33 分子軌道エディターの「New MOs」「Calculation」「Diagram」各パネル
それぞれのパネルで典型的な使い方を示しています(また必ずしもデフォルト設定ではありません)。
軌道占有数、電荷およびスピン多重度の修正

「Current Surface」ビューの右側には、表示が可能であれば分子のMOエネルギーおよび占有数を示すダイアグラムが表示され、最初はエネルギーの高い順に並べられています。αおよびβ電子は、青いボックスの中でそれぞれ上向きおよび下向きの赤い矢印で表示されています。対応するボックスの間をドラッグすることで、あるMOから別のMOへ電子を移動することが出来ます。MOボックスを右クリックすることで、MOから電子を追加あるいは消去することも出来ます。このようにMOの占有数を操作することで、分子の正味の電荷またはスピン多重度が適切に変更され、ChargeおよびSpin欄に反映されます。この2つの欄は直接編集することも可能で、変更はMOの占有数に反映されます。電荷を増やすと、HOMOに入っている電子から順に消去します。電荷を減らすと、LUMOから(あるいはHOMOの占有数が1であればHOMOから)順に電子を追加します。同様に、スピン多重度を増やすと最も高い2電子占有MOから最も低い仮想MOへ電子を移動し、またスピン多重度を減らすと最も低いエネルギーの1電子占有MOから最も高い1電子占有MOへ電子を移動します。最後に、電荷を奇数単位で変更すると、電荷の修正がスピン多重度にも影響を及ぼします。
「Calculation」パネルも軌道占有度の調整に使用できます。「Adjust Occupanciesポップアップには、以下の2つの選択肢があります:
- Aufbau:現在の電荷・スピン・波動関数の設定に沿って、軌道エネルギーが増加する順にMOを電子で満たします
- Original:適切であれば、電荷・スピン・波動関数を修正して、元のデータから読み込んだ軌道占有数あるいは計算した軌道占有数を復元します
軌道表示の制御
「Diagram」パネルでは、軌道図を制御します。「Arrange MOs」ポップアップにより、軌道をエネルギー、占有数、あるいはCIの順に並べることができます;後者はCASの活性空間(下記参照)を作成するのに適しています。「Degeneracy Thresholdでは、差がこれ以下であれば軌道エネルギーを等しいとみなす値を設定します(図34)。
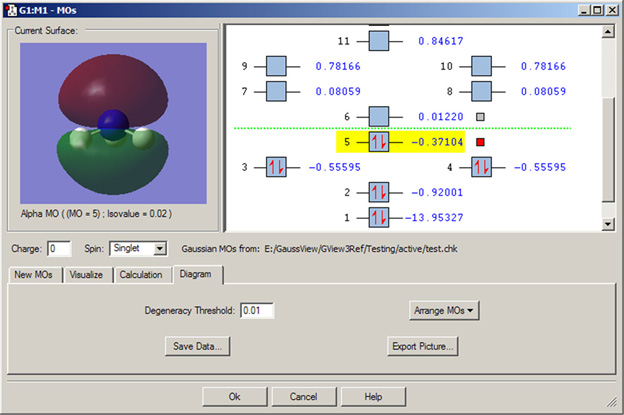
図34 分子軌道エディターでの縮退軌道
このNH3のMO表示では、軌道表示領域で縮退した軌道がどのように表されるかを示しています。ここでは、GaussViewが縮退したMOを検出できるように、Degeneracy Thresholdをデフォルト値である0から0.01に修正しています。
MOの作成と読み出し
「New MOs」パネルでは、Methodポップアップの設定により、MOデータの読み込みあるいは作成を行うことが出来ます。「Load Existing Chk File」を設定すると、checkpointファイルの場所を指定しLoadをクリックすることで、ファイルから軌道データを読み込むことが出来ます。
「Method」を「Generate New Chk File」に設定した時は、ModelおよびBasis Set領域で軌道を作成する計算モデルを設定します。「Model」と記された欄には初期MOの種類、局在化手法および波動関数の形式を設定します。このパネルの2行目の欄では、Gaussian計算に用いる基底関数を指定します。それぞれの欄にあるさまざまな選択肢についてのより詳しい情報は、Gaussian 03 User's ReferenceにあるGuessキーワードの議論を参照してください。「Generate」ボタンをクリックすることで、軌道データを生成するためのGaussianジョブが始まります(計算のタイプはGuess=Onlyです)。
軌道順序の再配置
「Calculation」パネルにある「Permutation List」には、現在の軌道の並び順が常に表示されています。この領域のポップアップには、軌道の並べ替えに関する3つの選択肢が提供されています:
- Occupancy (Alpha):α軌道の占有数に基づいて軌道を並べ替えます。ここでは電子が満たされていない軌道の下に満たされた軌道を並べ、かつ完全に占有された軌道を部分的に占有された軌道の下に並べます。各グループ内で、軌道はエネルギーの大きい順番に配置されます
- Occupancy (Beta):β軌道の占有数に基づいて軌道を並べ替えます(前の項目と同様)。非制限の波動関数についてのみ利用可能です
- CI:全ての占有軌道および全ての非占有軌道が、各グループでエネルギーの増加順に配置するように連続的に並べ替えます。この選択はCAS計算に適しています
その他のオプション
「Diagramパネルには以下の2つの操作があります:
- Save Data:MO図にある分子軌道の情報を、通常のテキストファイルに保存します:各軌道の番号、占有数、軌道エネルギーそしてfrozen軌道の数。非制限波動関数では、αとβの情報はブロックを分けて保存されます
- Export Picture:MO図のグラフィック画像を保存します。ファイル名とグラフィック書式の入力を促されます。
結晶エディターによる周期系の構築
「Edit=>PBC」メニューかもしくは「Crystal Editor」ボタンにより、PBCウィンドウが開きます。このツールは周期系の単位格子を指定するために用いられます。このツールの機能を利用するためには、Gaussianでの周期境界条件計算の実行方法に加え、周期系、空間群、および関連する用語についての理解が必要となります。この節では、この知識があることを仮定しています。
このウィンドウには、以下の5つのパネルがあります:
- Symmetry:単位格子の次元数、格子系および空間群の制限を指定します
- Cell:単位格子の形状の指定に加えて、セルを構成する原子の再配置および削減の制御も含みます
- View:ビューウィンドウ内で各方向にいくつ格子を表示するかの指定など、様々な項目の表示/非表示を制御します
- Contents:格子内の原子のリストが、組み込み型の原子リストエディターとして含まれています。ポップアップにより、結合を修正することも可能です
- Reduce:単位格子の次元数を(例えば、3次元系から面に)減らすことができます
大まかに見ると、周期系を構築する標準的な過程は以下の通りになります(より詳細な内容は、GaussViewのPBCチュートリアルを参照してください):
- Symmetryパネルを用いて、次元数と希望する空間群を指定します
- マウスもしくはContentsパネルを使って、格子に原子を追加します
- 不要な原子を削除します(例えば、必要な原子を追加するための近道としてフラグメントを用いた場合など)
- Cellパネルを用いて、単位格子の大きさを設定します
- 必要であれば、Contentsパネルを用いて結合を調節します
- 必要であればセルの構成原子を減らします
- Viewパネルを用いて希望する表示設定を指定します
以下のサブセクションで、個々のPBCウィンドウパネルでの制御について順に記します。
Symmetryパネル
図35に「Symmetry」パネルを示します。このパネルで、以下の設定を行います:
- Lattice Dimensions:系の並進対称性(すなわち周期性)の次元数を設定します:Oneはポリマーで、aは並進ベクトルです;Twoはシートで、aとbが並進ベクトル、Threeは結晶で、a、b、そしてcが並進ベクトルになります。
格子の次元数と、対応するセルの構成要素の次元数は、同じである必要はありません。例えば、ポリマー(1次元格子)を構成する単量体は、一般的に直線状ではありません。
- Minimum Lattice System:3次元セルの変更を、指定した格子系を保持するように拘束します。
- Enable Space Group Symmetry:空間群の対称性を監視し、空間群対称性を制限します。
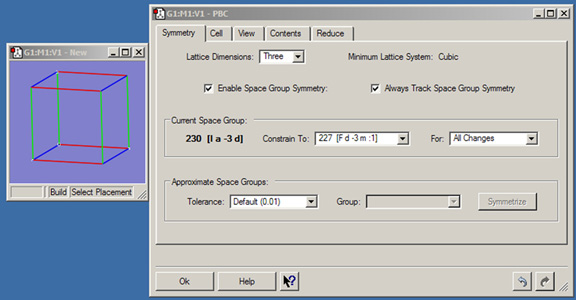
図35 PBC Symmetryパネル
このパネルでは、3次元の周期系を表示された空間群に制限するように設定していることを示しています。
- Always Track Space Group Symmetry:セルの中身の変更が完了した後にのみ対称性を計算せずに、セルの構成をインタラクティブに変更する(例えば、結合長スライダーを用いて連続的に)間ずっと空間群の対称性を監視します。
以下の操作はCurrent Space Groupパネルにあります:
- Constrain To:指定した空間群までは最低限保持するように変更を制限します。系が正確に属している全ての空間群を制限に用いることが出来ます。
- For:指定したタイプは最低限保持するように、空間群の対称性を強制します。Cell Changesは、セルの長さと角度についての変更を監視します。Contents Changesは、原子および結合の数、タイプ、または位置の変更を監視します。デフォルトはAll Changes(すなわち両方)です。
以下の操作はパネルのApproximate Space Group領域にあります:
- Tolerance:分率座標において、属している空間群を判定する近似の許容範囲を設定します。リストから選択したりこの欄に希望する数値を入れたりすることが出来ます。浮動小数点数(例えば0.25)あるいは分数(例えば1/4)で入力した値が入力可能です。
- Group:「Symmetrize」ボタンを押したときに対称化させる空間群を設定します。指定した許容範囲内で適用可能な全ての空間群がリストされます。
「Symmetrize 」ボタンを押すと、指定した群に系が正確に属するように対称化します。
Contentsパネル
「Contents」パネルには、通常の原子リストエディターとして機能するミニ原子リストエディターが組み込まれています(図36)。パネル上部には、以下の操作メニューがあります:
- Intercell Coordinate Changes:セル内の座標変化をどのように処理するかを特定します。選択肢としては、「Fix Contents’ Cartesian Coordinates」「Fix Contents’ Fractional Coordinates」「Fix Cell Parameters」があります。
- Bonds:セル内およびセル間の結合を再計算もしくは削除します。再結合は、距離を基準に判定します。このメニューにある様々な選択肢を図36に示します。
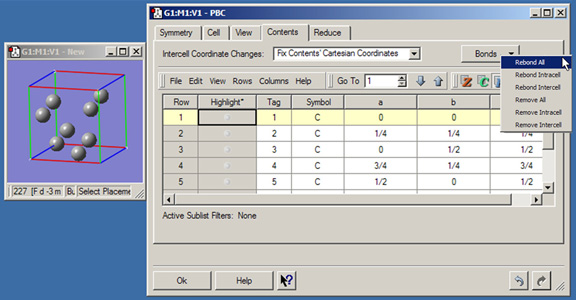
図36 PBC Contentsパネル
このパネルには、単位格子を構成する原子のリストが表示されています。ここには原子間を適切に再結合させることが出来るBondsボタンも含まれていて、主にCellパネル(図37)でパラメーターを修正した後に用います。
このパネルで原子を追加するには、原子リストエディターと同様の手順を用います。テーブルの最後の行(テーブルが空の場合は1行目)にAddボタンがあります。このボタンをアクティブにするためには、Symbol欄に元素タイプを入力します。新しく追加する原子に対して、残りの欄を使って分率座標を指定することも出来ます。追加の原子は、空間群の制限に一致させるためAddボタンをクリックしてはじめて追加されます。
Cellパネル
図37にCellパネルを示します。
「Cell Changes」ポップアップにより、セルパラメータを変更したときにセルの構成原子がどのように変更されるかを指定します。「Fix Contents’ Cartesian Coordinates」を選択した場合、セルパラメータ変更後のセルを構成する原子とそれらのデカルト座標は変化しません。
「Add/Remove Atoms」チェックボックスの設定は、操作に対して以下のように影響します:
- チェックしてない場合、パラメータを変更した後のセルを構成する原子とそれらのデカルト座標は、変更前と同じになります。例えば、セル内に分率座標(1/2, 1/2, 1/2)の原子が1つあり、aの長さを2倍にした場合、変更後のセル内は原子数は1のまま、分率座標は(1/4, 1/2, 1/2)になります。
- チェックしている場合、パラメータ変更後にセルの境界内にある全ての原子を、変更後のセルの構成原子として定義します。例えば、セル内に分率座標(1/2, 1/2, 1/2)の原子が1つありaの長さを2倍にした場合、変更後のセル内は原子数は2と増え、分率座標は(1/4, 1/2, 1/2)および(3/4, 1/2, 1/2)となります。
「Cell Changes」を「Fix Contents’ Fractional Coordinates」に設定した場合、パラメータ変更後のセルの構成原子とそれらの分率座標は変更前と同じになります。例えば、セル内に分率座標(1/2, 1/2, 1/2)の原子が1つありaの長さを2倍にした場合、セルの構成原子は1つのままでありその分率座標は(1/2, 1/2, 1/2)のままになります。多くの場合、2つのオプションのうち、こちらのオプションの方がより有用でしょう。
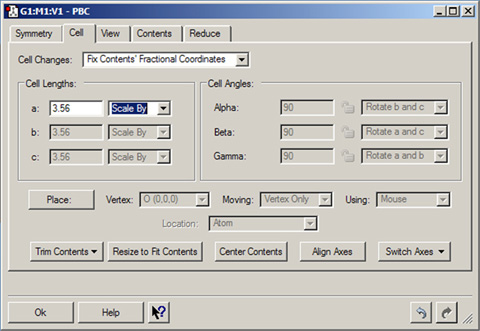
図37 PBC Cellパネル
このパネルは、単位格子のサイズを変更するのに用いられます。変更できない欄の値は、構造を制限する空間群によりあらかじめ定められたものです(図35のSymmetryパネルを参照してください)。
「Cell Lengths」と「Cell Angels」領域にある操作により、セルの構造を調整します:
- a, b, c:表示されているセルベクトルの長さを設定します(単位はオングストローム)。
- Scale by:セルベクトルの長さを正数でスケールします。
- Alpha, Beta, Gamma:他方のセル軸の間(すなわち、Alpha欄はbセル軸とcセル軸との間)の角度を設定します(単位は度)。「Rotate」欄で指定された内容に対応して、他方の2つのセル軸のうちの1つあるいは両方を回転することで、指定した角度に変更されます。非直交のセルであっても、角度を変更するために用いられる回転操作は他の2つの角度を変更しないように制限されます。
- 錠アイコン:ロックされている場合、角度を現在指定されている値に固定します。この角度は、頂点を配置している間もその値のまま拘束されます。
- Rotate:角度を変更した場合に回転する軸を指定します。他方の2つの軸のうち、どちらか1つあるいは両方の軸のいずれかが選択肢となります。
図38に、図37のようにセルの長さを指定してContentsパネルで再結合(図36参照)させた後のGaAsの単位格子を示します。
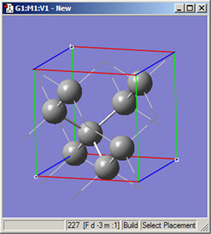
図38 修正し再結合したGaAs単位格子
図37および36の操作をした後の表示です。
パネル内の次の2行にある欄では、セルの頂点の配置(単位格子の境界を決める)を調整します。
- Place:セルの頂点の配置変更を可能あるいは不可能にします。
- Vertex:設置するセルの頂点を指定します。
- Moving:頂点のみを動かすか、あるいはセル全体を動かすかを指定します。
- Using:頂点の配置を、ビューウィンドウでのマウス選択で行うか、あるいは分率座標にするかを指定します。
「Using」を「Mouse」に設定した場合、ビューウィンドウでのマウスを用いた頂点の配置についてポップアップでその形式を指定します。ポップアップ内のAtom」は、クリックした原子上に頂点を置くことを意味します。「Between Two Atoms」は、マウスでクリックした2原子間の中間に頂点を置くことを意味します。また「Any Point 」は頂点を空間内のクリックした点に置くことを意味しています。
「Using」を「Coordinates」に設定した場合、分率座標を指定するための欄が現れます。Applyボタンをクリックすることで、指定した座標に頂点が配置されます
パネル底部にある5つのボタンには、以下の用途があります(3次元セルを例として用います):
- Trim Contents:指定した方法を用いてセルの構成原子を変更します。「Delete All Atoms Outside Cell」が選択されている場合、セル境界の外にある全ての原子は削除されます。「Translate All Atoms Inside Cell」が選択されている場合、セル境界の外にある全ての原子はセル境界内の等価な位置に移されます。余分な原子は削除されます。
1つ以上の分率座標が0未満もしくは1以上である場合、その原子はセル境界の外にあります。
- Resize to Fit Contents:セルの構成に合わせてセル長を変更し、構成原子を中央に移動します。セルの各辺に0.5オングストロームのクッションを追加します。
- Center Contents:セルの中心(1/2, 1/2, 1/2)が構成原子の核電荷の中心と一致するようにセルを移動します。
- Align Axes:セルの原点と軸をデカルト座標の原点および軸にそろえます。最初に、セルの原点をデカルト座標の原点に移します。次に、c軸とデカルトのZ軸がそろうようにセルを直角に回転させます。そして、b軸とデカルトのY軸がそろうようにセルを直角に回転させます。最後に、a軸とデカルトのX軸がそろうようにセルを直角に回転させます。
- Switch Axes:指定したペアのセル軸を交換します。例えば、a <–> bを選択した場合、aベクトルがbベクトルになりかつその逆も同じです。
Reduceパネル
図39にReduceパネルを示します。このパネルには以下の項目が含まれています:
- h, k , l:a、b、もしくはc軸(それぞれ)に沿った縮小格子切片のためのミラー指数を設定します。
- n(h), n(k), n(l):a、b、もしくはc軸に沿った縮小格子切片のための倍数を設定します。
- Highlight Reduced Lattice:ビューウィンドウ内に操作中の縮小格子を表示します(図39参照)。
- Reduce Lattice:指定したミラーパラメータに従い、格子を一次元に縮小します。
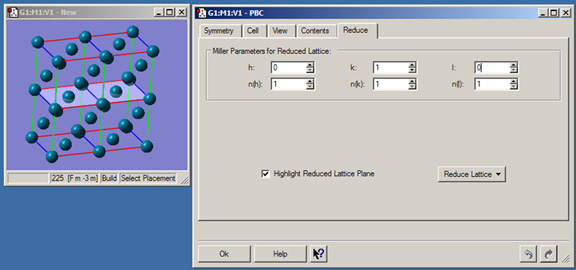
図39 PBC Reduceパネル
このパネルは、単位格子をより低い次元数に変換するために使用します。ここでは、パラジウムの3次元セルを2原子幅の面に縮小しています。明るい色の平面は切断面を示しています。
Viewパネル
図40にViewパネルを示します。このパネルには以下の機能が含まれています:
- a, b, c, -a, -b, -c:指定した軸に(表示されている、正あるいは負の方向に)沿って表示するセルの数を設定します。この数字の用途は表示のみで、Combineボタンを押さない限り、単位格子の構成原子に変更はありません。
- Combine:表示されている全てのセルを一体化して、より大きな「スーパーセル」にします。
- Show Reference Cell Edges:参照セルの辺および頂点を表示します。
- Show Replicate Cell Edges:全ての複製セルの辺および頂点を表示します。
- Show All Boundary Atoms:セル境界上にある全ての原子を表示します。
- View Along:指定した系のデカルトもしくはセル軸を、コンピューターのモニター面に垂直な軸にそろえます。
- Replicate Contents Display:複製セルにある原子および結合をどのように表示するかを指定します。選択肢としては、通常表示・光沢のない表示・環境設定「Display Format」の「Molecule」パネルで定義されたONIOMレイヤーの各表示形式そして非表示があります。
- General Positions表示している(制限された)空間群での等価位置が(分率座標で)リスト表示されます。「Display」ポップアップにより、空間群での一般等価位置に対応する非対称単位をどのように表示するかを指定します。
- Special Positions:表示している(制限された)空間群での特殊等価位置が(分率座標で)リスト表示されます。「Display」ポップアップにより、空間群での特殊等価位置をどのように表示するかを指定します。
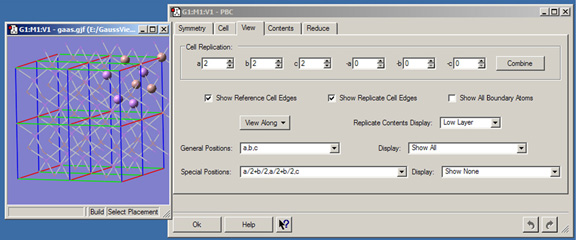
図40 PBC Viewパネル
このパネルでは、ビューウィンドウ内にいくつのセルを表示させるかを指定します。このビューウィンドウには、ヒ化ガリウムの3次元構造を示しています。各方向に2つのセルを表示しています。複製セルの構成原子は、Lowレイヤー表示形式(複製セルのデフォルトの表示モード)を用いて表示されています。
フラグメント配置動作の設定
GaussViewコントロールウィンドウのフラグメント領域の上にある小さな三角形をクリックすることで、いくつかのオプションを表示できます。これにより、新しいフラグメントを存在している分子構造に対してどのように加えるかを指定することが出来ます。ポップアップメニューMode により、Custom設定の指定や以前使用したデフォルトではない設定(Old、修正は不可)といった選択肢から選択することが出来ます。項目を変更するには、Customを選択する必要があります(初期状態では全ての欄が変更不可)。これら3つの設定グループに対する設定値は、環境設定の「Building」で指定することが出来ます。
この領域には2つのパネルがあります。図41に示したBeforeパネルでは、ある構造に対してフラグメントを追加する際の制限を設定します。Afterパネル(図42)では、フラグメントを追加した後の構造をどのように扱うかを指定します。
ここでは、このパネル内のオプションを理解するために必要な予備定義をいくつか示します:
- 「target atom」とは、マウスのクリックにより選択した原子のことです。
- 「hot atom」とは、2つのフラグメントをつなぐ時に「target atom」と置き換わる、新規フラグメント内の原子のことです。
- 「terminal atom」とは、1つの原子としか結合していない原子のことです。
- 「terminal valence atom」とは、水素原子または1つの原子としか結合していない開殻原子価のことです。
- 「join-bond」とは、「hot atom」と「terminal atom」との間の結合のことです。これらの結合は、フラグメントを「target atom」に結合させる過程で取り除くことが出来る結合です。フラグメントを「target atom」とつなげる際には、「target atom」との結合に対応した「join-bond」が1つフラグメント内に必要です。
フラグメント配置Beforeパネル内の項目を以下に示します:
- Target and Fragment Join-Bond Types Must Match:フラグメント内で選択した「join-bond」は「target atom」との結合のタイプに一致している必要になります。この項目を有効にすることで、過剰な置換を回避できます。
- Fragment Join-Bonds Can Be Any Terminal Bonds:通常は、水素原子または開殻原子価のみが「join-bond」を構成します。このオプションを「on」にすると、「terminal atom」も「join-bond」を形成する候補となります。例えば、ホルムアルデヒドの酸素原子をエチレンとのjoin-bondとして用いることが出来ます。なぜなら、結合のタイプが一致(2重結合)しているからです。
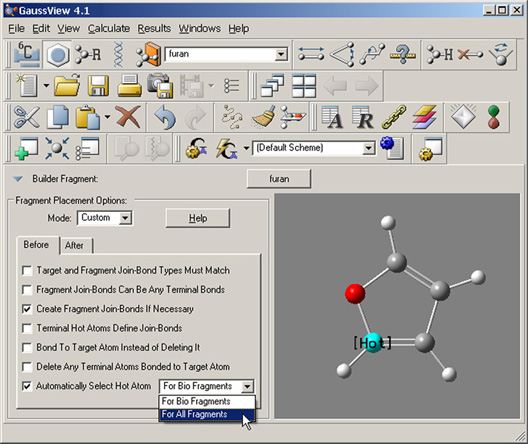
図41 フラグメント配置制限
このウィンドウ内の項目で、フラグメントをすでにある構造につなげる際の制限を設定します。この図では、デフォルトの値を示しています。
- Create Fragment Join-Bonds If Necessary:このオプションにより、ほとんどのフラグメントをあらゆる対象につなげることが可能になります。ある選択した対象に対して十分なjoin-bondがフラグメントにない場合、必要条件を満たすように原子価を追加します。重複しますが、フラグメントと対象を「target atom」でつなぐ場合、「join-bond」と「target atom」は削除されます(デフォルトではon)。
- Terminal Hot Atoms Define Join-Bonds:このオプションでは、「terminal atom」を「hot atom」として選択することを容易にします。ほとんどの「terminal atom」は「join-bond」を持っていませんので、このオプションでは1つの結合を「join-bond」として用います。しかし、この結合は消去されません。これは、2つ以上の終端結合をもつ中心原子があり、結合させるのに用いる終端結合を明確に選択したい時に有効です。
- Bond to Target Atom Instead of Deleting It:このオプションでは、フラグメントと「target atom」自身を結合させ、join-bondのみを消去します。
- Delete Any Terminal Atoms Bonded to Target Atoms:余分な終端結合を削除します。
- Automatically Select Hot Atom:この項目は「biological fragments」での使用を目的に作られたものです(デフォルト設定)。この設定によりGaussViewでは「hot atom」が動的に決められます。残基を追加するのにふさわしい主鎖の生成により、アミノ酸残基などを作成することが容易になります。

図42 フラグメント配置後の構造オプション
このパネルのオプションでは、ある分子構造にフラグメントを追加した後に自動的に実行される動作を選択することが出来ます。この例の設定がデフォルトです。
Afterパネルにある項目は以下の通りです:
- Use Fragment Join-Bond Type For New Bond:通常、フラグメントと対象とをつないでいる結合のタイプは、元の対象となる結合により決められます。このオプションをonにすると、代わりにフラグメントの結合タイプが用いられます。このオプションは、Match Bond Typesオプションがoffの時に便利です。
以下の3つの項目は、「target atom」側での電子数の収支調整をアクティブにします。しかし、非末端原子には影響しません。また、必要な原子価を満たすために結合タイプを減らしたり上げたりも行いません。
- Delete Terminal Valence Atoms to Satisfy Valency:「target atom」側での過剰な原子価を減らすために、末端原子価原子を削除する設定にします。
- Delete Terminal Atoms to Satisfy Valency:過剰な原子価を減らすために、末端原子を削除する設定にします。
- Add Hydrogens to Satisfy Valency:必要な原子価を満たすように自動的に水素原子を追加します。
残りのオプションは自動構造整形操作を有効にします。これらの機能は読んで字の通りです:「Optimize Dihedral Angle About New Bond」、「Apply “Clean” Around Hot Atoms」、そして「Apply “Clean” to Whole Structure」が有ります。
|
