分子ファイルと画像の操作
分子表示のカスタマイズと操作
GaussViewには、ビューウィンドウの表示をカスタマイズするための多くの機能が含まれています。これらの機能は、「View」メニューおよびいくつかはツールバーのアイコンから呼び出せます。「View」メニューは、ビューウィンドウや分子が表示されているウィンドウの何も無い部分でマウスの右ボタンをクリックする事でアクセス可能です。
分子のセンタリング
「Center」ボタンあるいは「View=>Center」メニューを選択すると、操作中のビューウィンドウの画像が中心に移動されると同時にサイズ調整されます。
任意の表示項目
以下の「View」メニューオプションを、ビューウィンドウの表示を切り替えるのに使用します:
- View=>Hydrogens:水素原子を表示(初期設定ON)
- View=>Dummies:ダミー原子が有れば表示(初期設定ON)
- View=>Labels:原子の通し番号の数字ラベルを表示(初期設定OFF)
- View=>Symbols:各原子の元素記号を表示(初期設定OFF)
- View=>Bonds:原子間の結合を表示(初期設定ON)
- View=>Cartesian Axes:X, Y, Z軸を表示(初期設定OFF、図43)
始めは、軸の原点は任意の位置に置かれます(原子1が原点となっている事も有ります)。分子が修正されたとしても、軸は固定されたままです。「 Edit=>Reorient」メニュー項目を使用すれば、原点を分子の重心へ移動できます。
- View=>Stereochemistry:不斉中心が存在していれば、立体配置を表示(初期設定OFF)
- View=>Positioning Tools:ビューウィンドウに位置調整用ツールバーを表示(初期設定OFF)
これらの項目のデフォルト値は、「View」メニューの「Display Format」で表示されるダイアログの「General」パネルあるいは「Preference」の「Display Format」画面で設定できます。また「Display Format」設定の「Text」パネルの「Exclude View Labels」と「Exclude View Symbols」項目で、水素原子あるいは水素原子と炭素原子のラベルや記号表示だけを除外する事が可能です。デフォルトでは、除外設定はされていません。さらに、このパネルでは、文字ラベルのサイズを「Size」欄で調整できます。
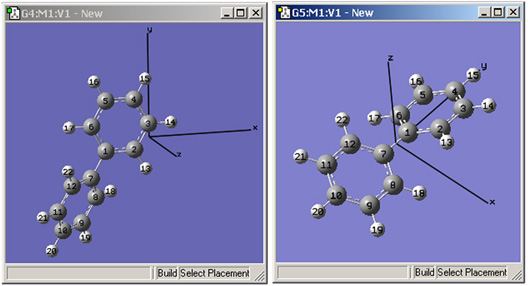
図43 デカルト座標軸の表示
これらのウィンドウは、GaussViewの「Cartesian Axes」表示オプションを示しています。
左のウィンドウは、軸の最初の位置です(任意の場所に表示されています)。
右は、「Edit=>Reorient」メニュー項目使用後の新しい位置を示しています。
分子の位置調整ツール
図44に、分子の位置調整用ツールバーを示しました。これは、「View=>Positioning Tools」メニュー項目を選択して表示できます。

図44 分子の位置調整用ツールバー
以上のツール全てにおいて、ビューのX, Y, Z方向は、ウィンドウの水平、垂直、奥行の各方向に一致します。
分子のX, Y, Z方向は、デカルト座標軸で示されます。
各コントロールを、ツールバーの左から右へ順番に、以下で説明します。
- 「Quick」ポップアップメニューには、分子を各軸の中心に移動するための項目や、分子の軸をビューの軸にそろえる項目が有ります。例えば、「Center」の「X」を選択すると、分子をウィンドウの中心へ移動し、ウィンドウのX軸(水平軸)に合わせます。また、「Mol X -> View Z」項目は、分子のX軸をビューウィンドウのZ軸に合わせます。他の項目も、同じように働きます。
- 二番目のポップアップメニューでは、「Apply」ボタンを押した時に位置調整を行うため分子を回転させるのか平行移動させるのかを指定します。これに続く次の2つも、操作を調整するための物です。
- 二番目のポップアップメニューで「Rotate」を選択すると、隣に「Around」と表示されます。そこでは、分子を回転させる際の軸を指定します。二番目のポップアップメニューで「Translate」を選択した場合には、隣の表示は「Along」となります。
- 「By」欄には、回転や平行移動の量を設定します。
- 最後のスライダーを使用して、移動量を指定することもできます。スライダーを動かすのと同時に、一および二番目のメニューで設定した様式で分子が動きます(この場合、「By」欄の数値は無視されます)。
分子の表示フォーマット
「Display Format」ボタンか「 View=>Display Format」メニュー項目を使用すると、「Display Format」ダイアログを開くことができます。このダイアログの各パネルは、「Preferences」の「Display」にも含まれています。ここでは、「General」「Text」「Molecule」各パネルの説明をします。「Surface」パネルに関してはここ以降で説明します。
「General」パネル
「General」パネルの各項目で、分子表示の品質を調整できます。原子への上述したような追加表示項目やフォグによるぼかし効果の調整もできます(図45)。
このパネル上部のオプションは、ビューウィンドウ内の分子がマウス操作で動いている時とそうでない時の、表示品質を規定します。動作スピードの遅いコンピューターでは、動かしていない時の「Stationary Object Display」の設定はスライダーを「Realistic」側にしておいて、動かしている時の「Moving Object Display」のスライダー設定を「Fast」側にすれば操作しやすいと思います。
「Smooth」チェックボックスは、ディスプレイ内の表示のスムージングを有効にしたり無効にしたりするための物です。「Spotlight」チェックボックスは、スポットライトのシミュレーションを切り替えるための物です。
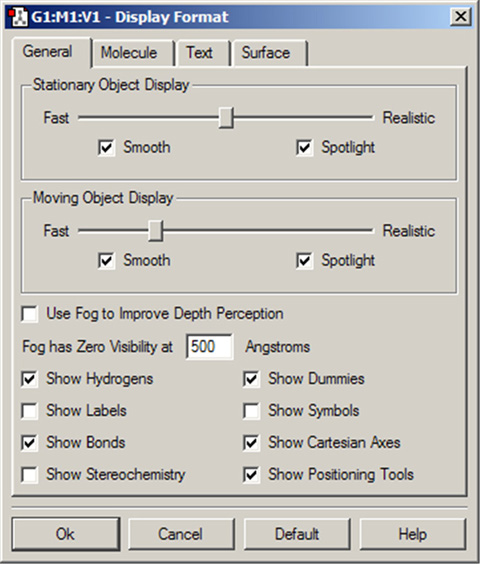
図45「Display Format」の「General」パネル
「Use Fog to Improve Depth Perceptionチェックボックスは、ビューウィンドウの奥に離れている原子を薄い色にして見えなくしていき、手前の原子をよりはっきりとさせるための物です。「Fog has Zero Visibility at」欄は、原子が完全に見えなくなる深さを調整するための物です(図46)。
図46の例では、左側はフォグの効果を切っており、右側はフォグを有効にしています。手前の原子が奥の物よりも、よりはっきりと表示されているのが分かると思います。
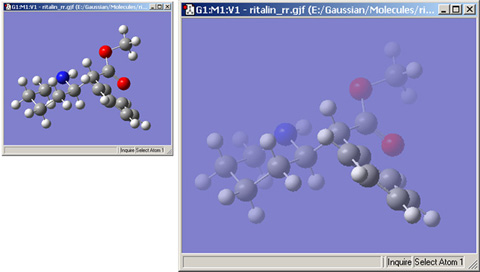
図46 フォグによるぼかし効果
両者を比較すると、フォグによる効果が分かります。
「Molecule」パネル
のパネルでは、原子と結合の表示形式を設定できます(図47)。三つのポップアップメニューで、ONIOMレイヤーの各層の表示形式を設定します。もしもONIOM機能を使用していない場合、「High Layer」の設定が使用されます。
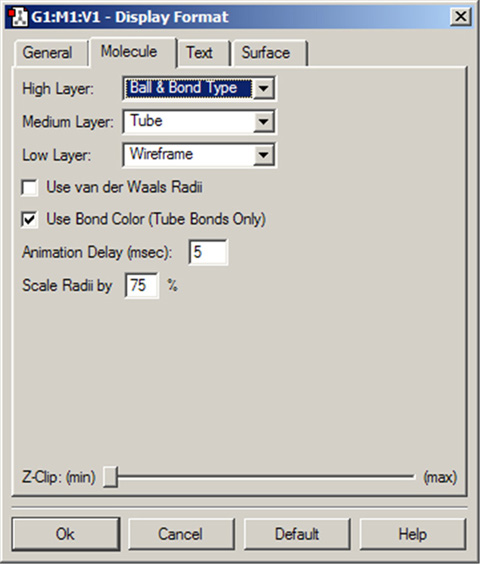
図47「Display Format」の「Molecule」パネル
分子の表示の様々な調整を行います。
レイヤの表示設定は、ONIOM以外にもPBC単位格子の境界での原子表示などにも使用されます。
以下の表示形式がサポートされています:
- Ball & Stick:分子がボール&スティックモデルで表示されます。全ての結合は、1本の棒で表されます。デフォルト設定。
- Ball & Bond Type:分子はボール&スティックモデルで表示され、単結合は1本の棒で多重結合は多重度の数の棒で表されます。芳香族の場合、点線が追加されます。
- Tube:分子が、チューブで表されます。結合タイプは無視され、原子種はチューブ上の色バンドで示されます。
- Wireframe:分子が、細い線の組み合わせで表示されます。原子タイプで色分けされ、結合タイプは複数の線で表されます。
- None:何も表示されません。多層モデルの際のみに使用します。
「Use Vanderwaals Radii」チェックボックスと「Scale Radii」欄を使用して、ビューウィンドウ内の原子の大きさをvan der Waals共有結合半径の比率で指定できます。比率を150%-200%にすると、空間充填モデルのような表示にできます(図48)。
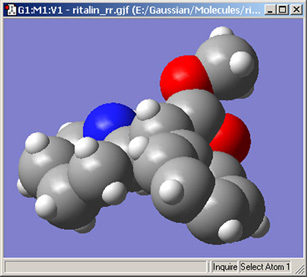
図48 CPK様表示
「Z-Clip」スライダーを使用して分子の手前の表示を消去し、内部を覗くことができます。
「Use Bond Color」チェックボックスで、チューブ表示における表示色を全て原子の色にするのか一部だけにするのか切り替えできます(図49)。
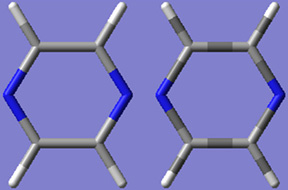
図49 チューブ表示形式
ここでは、ピラヂンをチューブ形式で表示してあります。
左は「Use Bond Color」チェック無しで、右はチェックをした表示です。デフォルトは、左側です。
「Text」パネル
Display Format」ダイアログの「Text」パネルでは、フォントと原子ラベルと記号を設定できます。上で説明したように、ラベル表示を除外する原子の設定もできます(図50)。
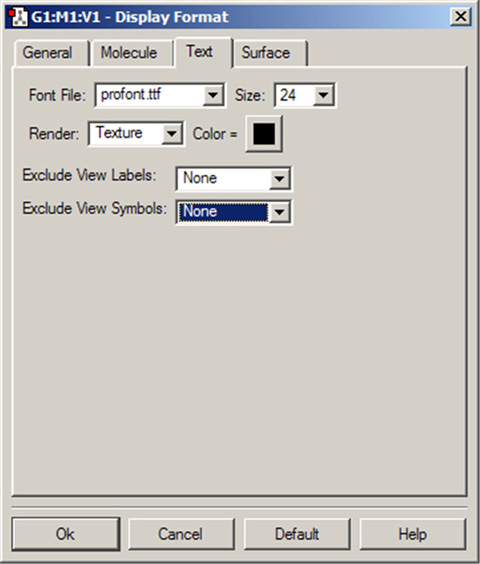
図50「Display Format」ダイアログの「Text」パネル
このパネルでは、原子のテキストラベルの様々な設定を行えます。
「Font File」メニューを使用して、使用するフォントをGaussViewにより提供されているいくつかのフォントファイルから選択することができます。「Size 」欄では相対的なテキストサイズを指定できます。テキストサイズは、分子を拡大縮小するのに合わせて変化します。「Render」メニューでは、テキストのレンダリング手法を選択できます。「Color」では、テキストの色を指定します。
表示色のカスタマイズ
「Preferences」の「Colors」では、ビューウィンドウの背景・原子・結合・表面・振動ベクトルの各表示色を調整できます。ダイアログを、図51の左側に示します。
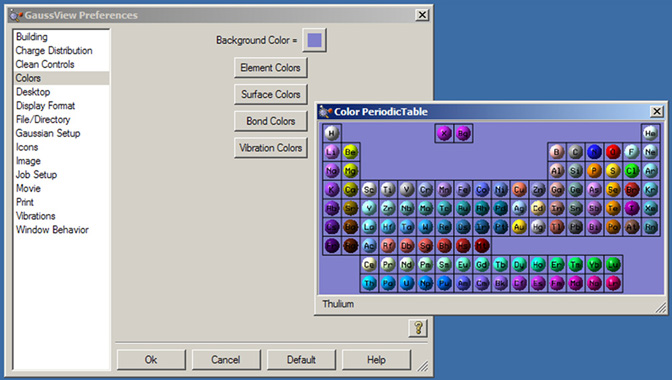
図51 GaussViewの色の調整
「Preferences」の「Colors」ダイアログを左に、「Element Colors」設定を右に示しています。
「Preferences」の「Colors」ダイアログでは、以下の項目の色調整ができます:
- Background Color:ビューウィンドウの背景色。修正するには、右のカラーチップをクリックします。
- Element Colors:元素単位での色分け。このボタンをクリックすると、周期律表が表示され、各元素のデフォルトの色が示されます(図51)。元素をクリックすると、デフォルトの色設定を変更できます。
- Surface Colors:表面の色を設定(後で説明します)
- Bond Colors:各表示タイプでの、結合の色を設定。ダイアログは図52と似たような感じで、選択した色が球で示されます。原子ではなく、結合に使用される色を設定するダイアログですので、混乱しないようにしてください。
- Vibration Colors:振動の表示における、双極子導関数や変位ベクトルの色を設定。ここでも図52のようなダイアログが表示されますが、原子ではなく表示されるベクトルの色が変更されます。
色の指定
図52に示したダイアログで、単一色の表面を設定します。正と負のローブを持つ表面は、それぞれの色を設定します。図は、分子軌道表面の設定を行っています。他の色を設定するダイアログも、これと同様の構成になります。
ここで示したダイアログの例では、上部の項目で表面のタイプと表示方法を指定できます。他のダイアログでは、これらは適切に置き換わります。最も重要な項目が、ダイアログの中央に表示されています。「Scheme」ポップアップで、オブジェクトに反映させる色づけのタイプを設定します。グラデーションの色の塗り方を「1-Color Shaded」「2-Color Shaded」「3-Color Shaded」「4-Color Shadedから選択することにより、指定した色(1-Color Shadedの場合)あるいは複数の指定した色の混合(2~4-Color Shadedの場合)で陰影の色を指定できます。ただ、色の指定は少ない方が、グラデーションはきれいになります。「1-Color Flat」の設定では、三次元的な表現は行わず、指定した色で塗りつぶされます。
ここに表示されているカラーチップをクリックすれば、色を変更できます。ローブが2つ有る表面の場合、ダイアログの左右に2組のカラーチップが表示されます。もしも「Use RGB Selectorr」チェックボックスが選択されていると、下に表示されるスライダーを使用してRGBの各値を調整して色を変更できます。選択されていなければ、システムの色指定ユーティリティが起動されます。
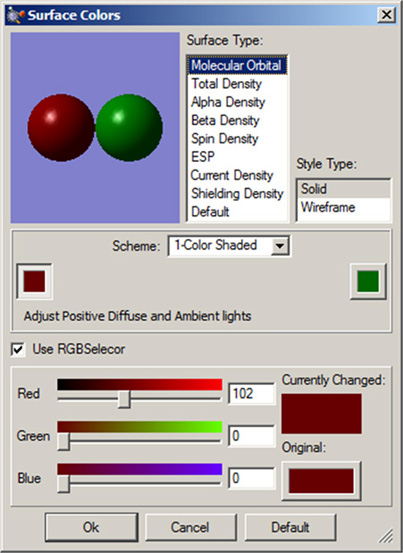
図52 表面の色を指定する設定パネルs
このダイアログで、表面に使用する色を設定できます。
表示の追加と同期
「View=>Add View」メニュー項目あるいは「Add View」ボタンを使用すると、現在のモデルに対する新しいビューウィンドウを追加できます。このビューは、デフォルト設定から始めて個別に調整できます。
「View=>Synchronize」メニュー項目を使用すると、同じモデルの別のビューをリンクして、マウスによる回転・平行移動・拡大縮小などの操作を同期できます。この設定は、同期させたい各ビューウィンドウでそれぞれ行う必要があります。同期は、他の設定に影響を与えませんが、構造に修正が加えられた場合は、自動的に同期されます。
ビューウィンドウの選択
「Windows」メニューには、ビューウィンドウの表示を制御する項目が含まれています。分子グループの全てのビューと付随するダイアログは、「Windows=>Molecule Groups」メニューに表示されるサブメニューの該当する項目のチェックを入れたり外したりする事で、表示させたり隠したりできます。ここのビューウィンドウは、「Windows」メニュー下部の対応する項目を使用して、表示させたり隠したりできます。このメニューでウィンドウを選択すると、それがアクティブビューとなります。ここで、隠すという事は閉じる事とは違うという事を理解しておいてください。画面のスペースを管理するだけの操作です。また、ウィンドウの最小化とも違い、隠したウィンドウはドックやタスクバーにアイコン表示されなくなります。
「Windows=>Molecule Groups」サブメニューの「Show All」と「Show None」は、全てのモデルグループそれぞれのウィンドウを表示したり隠したりします。これに関連して、「Windows=>Minimize All」「Windows=>Restore」「Windows=>Restore All」の各メニュー項目は、それぞれウィンドウの最小化、アクティブビューの表示、最小化した全てのビューウィンドウ(隠したもの以外)の表示を行います。「Windows=>Close」と「Windows=>Close All」は、それぞれ作業中のウィンドウあるいは全てのビューウィンドウを閉じます。
「Windows=>Previous」と「Windows=>Next」メニュー項目(および対応する「Previous」と「Next」ボタン)は、順に前あるいは次のウィンドウをアクティブにします。
最後に、「Windows=>Cascade」と「Windows=>Tile」メニュー項目(および対応する「Cascade」と「Tile」ボタン)を使用して、全てのウィンドウを重なった状態や可能な限り多くのウィンドウが画面上に表示されるように大きさを調整して並べ替えることができます。後者の場合、可能なら全てのウィンドウを表示します。
分子とファイルの操作
ここでは、分子と分子グループに関連する操作について説明します。外部ファイルの操作に関しても言及します。
ビューウィンドウへの分子のペースト
「Edit=>Cut」と「Edit=>Copy」メニュー項目と対応する「Cut」と「Copy」ボタンを使用して、システムのクリップボードへ現在のモデルの全ての構造をカットあるいはコピーできます。その際、モデルでの原子の選択状況は無視されます。
「Edit=>Delete Molecule」メニュー項目と「Delete Molecule」ボタンを使用して、現在のモデル内の構造を削除できます。それらは放棄されて、どこにもコピーされません。

クリップボードから分子をペーストするには、いくつかオプションがあります。「Edit=>Paste」メニュー項目では、3つの項目を含むサブメニューにアクセスできます。これらは、「Paste」ボタン横の小さな下向き三角形をクリックしてもアクセスできます。この三角形ではなく「Paste」ボタンをクリックした場合は、「Add to Molecule Group」項目が実行されます。
「Paste」のサブメニュー項目は以下のようになります:
- Edit=>Paste=>Add to Molecule Group:使用中の分子グループに新しいモデルを作成し、クリップボードの構造を置きます。
- Edit=>Paste=>Replace Molecule:操作中のウィンドウの構造を削除し、代わりにクリップボードの構造を置きます。
- Edit=>Paste=>Append Molecule:クリップボードの分子を、使用中のモデルに別のフラグメントとして追加します。
Windows環境下でのファイルのドラッグ&ドロップ
Windows環境下では、エクスプローラーウィンドウなどからファイルをビューウィンドウにドラッグ&ドロップして、操作中の分子グループに追加できます。左マウスボタンを押しながらドラッグ&ドロップした場合、各ファイルの中身が読み込まれ、分子グループに追加されます。
デフォルトではファイルの中身が操作中の分子グループの新しい分子として配置されますが、右マウスボタンを使用してドラッグ&ドロップした場合、コンテキストメニューが開き以下のオプションを選択できます:
- Add all files to this molecule group:ファイルは、使用中の分子グループに追加されます(デフォルト)
- Append files to this molecule:ファイルの中身は、操作中の分子に追加されます(使用中の分子グループに新しいフレームは作成されません)
- Separate new molecule group for each file:各ファイル用に新しい分子グループが作成され、操作中の分子には影響しません
- Single new molecule group for all files:新規に1つの分子グループが作成され、各ファイルはその中の分子(フレーム)として配置されます。使用中の分子グループには影響しません
ファイルを1つだけドラッグした場合は動作が変わり、最後のオプションは表示されません。
モデルの追加
「File=>New」メニュー項目から、以下の2つのサブメニューにアクセスできます:
- Create Molecule Group:新しく分子グループが作成され、その中のモデルに対応する新しいビューウィンドウが表示されます
- Add to Molecule Group:操作中のモデルグループに新しくモデルが追加され、対応するビューウィンドウが表示されます
どちらの場合も、新しいビューウィンドウは何も含んでいません。

これらの機能は、「New」ボタン横の小さな下向き三角形が表示されたボタンで実行できます。この三角形ではなく、「New」ボタンそのものをクリックした場合は、「Create Molecule Group」項目が実行されます。
ファイルを開く
「File=>Open」メニュー項目か「Open」ダイアログを表示させることができます。このダイアログの形式は使用しているコンピューターに特有のものとなりますが、図53のようにいくつかGaussView特有の項目が追加されています。
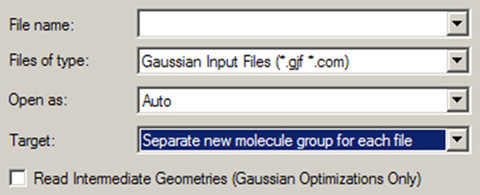
図53「Open File」ダイアログのGaussView特有の項目
オペレーティングシステムの通常の「Open File」ダイアログに、GaussViewが3つの項目と3つのチェックボックスを追加しています。
「Files of Type」ポップアップは、開くファイルの種類を指定します。これはまた、ダイアログのファイルリストにどのファイルを表示させるか選択するフィルターにもなります。表示されるファイルリストは、選択した拡張子を持つものだけになります。サポートされているファイル形式と拡張子は以下の通りです:
- Gaussian Input Files (.com または .gjf)
- Gaussian Output Files (.log または .out)
- Cube Files (.cub*)
- Gaussian Checkpoint Files (.chk)
- Gaussian Formatted Checkpoint Files (.fch*)
- Brookhaven PDB Files (.pdb または .ent)
- MDL Mol Files (.mol または .rxn または .sdf)
- Sybyl Mol2 Files (.mol2 または .ml2)
- CIF Files (.cif)
「All Files」を選択すれば、全てのファイルが表示されます。
「Open as」ポップアップは、実際の拡張子に関係なく、指定したファイル形式のものとして強制的にファイルを読み込みます。「Gaussian cube」ファイル以外の全てのサポートされている形式を選ぶことができます。デフォルト設定は「Auto」で、ファイル形式は拡張子のみで判断されます。
「Target」ポップアップでは、読み込んだ構造をどこに配置するのか以下から選択できます:
- Separate new molecule group for each file:各ファイル毎に新しく1つのメンバーからなる分子グループを作成して、それぞれに読み込みます(デフォルト)
- Single new molecule group for all files:新しく1つの分子グループを作成し、各ファイルをそこに追加して行きます。ファイルを1つだけ開く場合は、上のオプションと同様になります。
- Append all files to active molecule:全てのファイルの構造を、操作中の分子(使用中の分子グループ内)に配置します。
- Add all files to active molecule group:使用中の分子グループに、各ファイルが別々に新しい分子として追加されます。操作中の分子は、そのまま残ります。
通常は、ファイルに含まれる最初の構造だけが入力されます。そのため、Gaussian入力ファイルの最初の計算構造だけが取り出されます。しかし、「Read intermediate geometries (optimization only)」チェックボックスを選択した場合、GaussViewは指定した「Target」に対して、Gaussianの結果ファイルに含まれる全ての構造を別々のモデルとして取り出します。これは、構造最適化・IRC・ADMPとBOMDトラジェクトリー・ポテンシャルエネルギー面スキャンなどの構造最適化要素を含む各計算結果ファイルに適用されます。この機能は、「Append all files to active molecule」を選択した際には、適用されません。
最近使用したファイルや関連ファイルへのアクセス
「File=>Recent Files」メニューには、最近開いたファイルのリストが含まれています。ここで表示されるファイルの最大数は、「Preferences」の「File/Directory」内に有る「Maximum Recent Files」欄で指定できます。しかし、これらを指定してファイルを開くと、現在の「Open File」ダイアログの設定や以前開いたときの設定は無視され、1つの構造だけが読み込まれます。このファイルリストは、GaussViewを終了しても維持されます。
「File=>Related Files」メニューには、操作中の分子に関連するファイルのうち開くことができるもの(例えば、Gaussian出力ファイル、チェックポイントファイルなど)がリストされます。
「File=>Refresh」メニューは、操作中の分子を元のファイルから読み直し、分子をビューの中心へ移動し場合によっては再設定します。これは、操作中の分子を修正していない場合のみ使用できます。
開いているモデルの元ファイルの確認
「View File」ボタンを押すか「Results=>View File」メニュー項目を選択すると、使用中のモデルの元ファイルがテキストで開きます。ただし、分子構造が修正されていない事が条件となります。入力ファイルがGaussianチェックポイントファイルのようなバイナリーファイルの場合、GaussViewからformchkユーティリティが呼び出されて形式変換後、表示されます。
分子の保存
「File=>Save」メニューあるいは「Save」ボタンを使用して、操作中のモデルを外部ファイルあるいは一連のファイル群に保存できます。ダイアログには、使用しているシステムの「Save」ダイアログが使用され、GaussViewによりいくつかの項が追加されています(図54)。
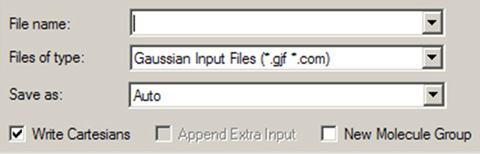
図54「Save」ダイアログ中のGaussView項目
GaussViewは、システムの標準「Save」ダイアログにいくつかの項目を追加します。分子は、いくつかの形式で出力できます。
それら項目は、以下の意味を持ちます:
- Files of type:フィルターとして働き、ダイアログのファイルリストに表示するものを指定します。
- Save as:指定した拡張子にかかわらず、選択した形式で分子を保存します。デフォルトは「Auto」で、拡張子を基にして形式を決定します。
- Write Cartesian Coordinates:チェックすると、分子構造はデカルト座標でファイルに保存されます。デフォルトは、Z-matrix形式です。
- Append Extra Input Data:これは、Gaussian入力ファイルを読み込んだモデルの場合に有効となります。ファイル内に追加入力が存在していた場合、これにチェックすると出力するファイルにもそれらが含まれます。
- Create New Molecule Group:チェックすると、保存した分子を含む新しい分子グループを作成します。
分子グループ内で操作中のモデルが保存されます。分子グループ内の全ての分子を1つのファイルに保存したり、1回の操作で一連のファイルにそれぞれ保存することもできません。
画像の印刷と保存
「File」と「Edit」メニューの様々なオプションやそれに関連するボタンを使用して、ビューウィンドウの中身を印刷したり画像ファイルとして保存したりできます。
印刷
操作中のビューウィンドウ内の画像は、「File=>Print」メニューを選択するか「Print」ボタンをクリックして印刷できます。通常の、システムの「Print」ダイアログが表示されます。
印刷は、「Preferences」の「Print」で設定できます(図55)。「Print」ダイアログの設定は、「Preferences」の設定よりも優先されます。
デフォルトでは、プリンターの解像度に合わせて画像の解像度も最大4倍まで変更され、背景は白色となります。画像は常に、操作中のサイズで印刷されます。画像を大きくしたい場合、代わりに「Enlarge Width and Height」を使用してください。「Gray Scale」が選択されていない限り、カラー画像が作成されます。
「Object Quality」スライダーにより、画像の品質を設定できます。スライダーを「Realistic」側へ移動すると、ファイルサイズが大きくなり画像ファイルを作成する時間が長くなります。出版用には、最大設定をお勧めします。
残りの設定は、低レベルでのグラフィックの扱いを設定します。「Generic Pixel Format」は、システムソフトウェアのレンダラーの使用を指示します。このオプションは、画像のキャプチャーや印刷に失敗した時にのみ使用してください。「Use Pixel Buffers」はOpenGLのピクセルバッファーの使用を指示します。環境によっては、グラフィック表示のパフォーマンスが上がる可能性があります。
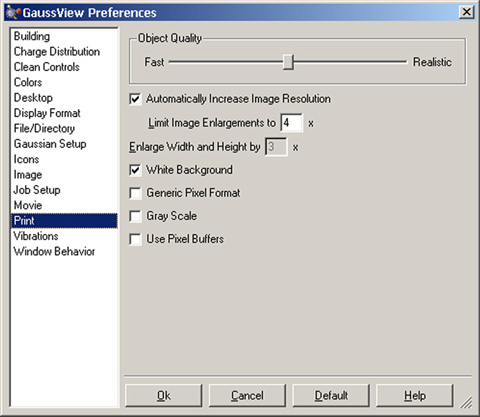
図55「Preferences」の「Print」パネル
このパネルでは、画像をプリンターの解像度に合わせて大きくする設定や、背景の設定ができます。
画像ファイルの保存
「File=>Save Image File」メニューか「Save Image」ボタンによって、画像をファイルに保存できます。これらを指定すると、追加の項目を含んだシステムの「Save」ダイアログが表示されます(図56)。
「Files of type」は、ファイルリストに表示するファイルのフィルターとして使用します。「Save as」は、作成する画像ファイルの種類を指定します。デフォルトでは、ファイルの拡張子で決定されます。使用できる画像タイプは、TIFF, JPEG, Vector Image, BMP, PNG, JPEG-2000, GIFです。通常は、高解像度印刷にはTIFF形式を、ウェブやプレゼンテーション用にはJPEGかPNGをお勧めします。
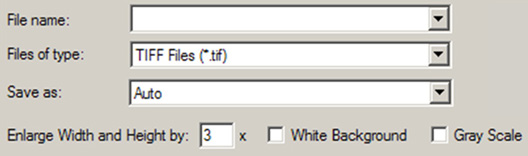
図56「Save Image」ダイアログ中のGaussView項目
これらの項目は、画像を保存する際にGaussViewによりシステムの「Save」ダイアログに追加されます。
このダイアログの他の項目は、以下の機能を持ちます:
- Enlarge Width and Height by:一定の倍率で画像を大きくすると同時に、解像度も高くします(デフォルトは3倍)。画像を作業中のサイズにおいて300 dpiで印刷するのなら、4倍を指定してください。画像の大きさをも大きくしたい場合は、この値をもっと大きくしてください。作業中の画像を300 dpiにおいて2倍のサイズにするには、8倍を指定してください。レンダリングの計算時間は、拡大率の二乗に比例して長くなりますので注意してください。
- White Background:白色の背景を使用します。この項目のチェックを外したら、ビューウィンドウの背景が使用されます。
- Use Gray Scale:画像をグレーに変換してから保存します。デフォルトでは、カラー画像が保存されます。
画像キャプチャと保存の設定は、「Preferences」の「Image」で設定できます(図57)。「Save」ダイアログの設定は、「Preferences」の設定より優先します。
このパネルの各項目は、以下の機能を持ちます:
- Object Quality:画像の品質を設定。スライダーを「Realistic」側に移動すると、作成する画像ファイルのサイズが大きくなると同時に作成時間も長くなります。
- Enlarge Width and Height by:デフォルトの画像拡大率を設定(上記参照)
- White Background:白色の背景を使用(デフォルト)。チェックを外すと、ビューウィンドウの背景が使用される
- Generic Pixel Format:システムのソフトウェアレンダラーを使用。このオプションは、画像キャプチャや印刷時にレンダリングの問題や失敗が有ったときのみ使用
- Use Gray Scale画像をグレーに変換。デフォルトでは、カラー画像を保存
- Use Pixel Buffers:OpenGLのピクセルバッファーを使用(環境によってはグラフィックスパフォーマンスが向上する可能性有り)
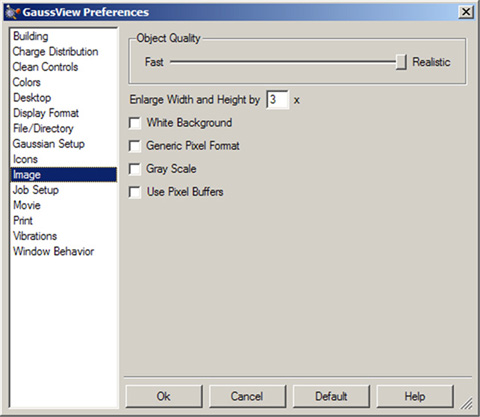
図57「Preferences」の画像キャプチャー設定
このパネルでは、画像キャプチャーのデフォルト設定を行います。「Movies」の設定に似ています。
動画ファイルの保存

「File=>Save Movie」メニューか「Save Movie」ボタンを使用して、操作中の分子グループの各フレームをアニメーションとして保存できます。このメニューのサブメニュー項目は、ボタン横の小さな三角をクリックしても選択できます。サブメニューの各項目は、以下の機能を持ちます:
- Save Movie File:複数のフレームを、一つのアニメーションとして保存。使用できるファイル形式はanimated GIFとMNGです。
- Save Movie Frame Files:分子グループの各フレームや分子を、別々の画像ファイルとして保存。このオプションは実際には、各分子を別々のイメージとして保存するためのショートカットとなります。この機能は、外部ソフトウェアを使用してばらばらのフレームファイルからアニメーションを作成したい場合にも便利です。
- Save Bothアニメーションと別々のフレームのファイルが両方とも保存されます。それぞれのファイル形式を指定するため、ダイアログが二回表示されます。
- ボタンの「Save Movie」アイコンの方をクリックした場合は、上記の「Save Movie File」が実行されます。
「Preferences」の「Movie」は、「Images」と同様です(図57)。
ファイル配置のデフォルト設定
「Preferences」の「File/Directory」を図58に示します。これにより、GaussViewに関連するファイルのデフォルトの位置を設定できます。
「Starting Directory」のポップアップで、以下の設定を行えます:
- Use previous work directory:直前に使用したディレクトリーを記憶して、次回にそれを使用
- Use launch directory:常に、デフォルトのディレクトリーとしてGaussViewを起動したディレクトリーを使用
- Specify:常に、指定したディレクトリーを使用
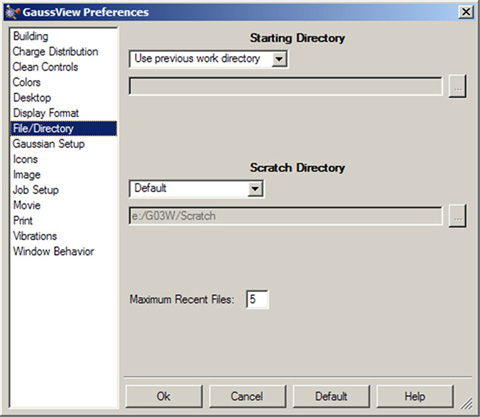
図58 デフォルトディレクトリーの指定
このダイアログでは、ファイルを開いたり保存したりするデフォルトのフォルダーの位置やGaussViewのスクラッチディレクトリーを指定します。また、「Recent File」リストに登録するファイルの数なども指定できます。
一時スクラッチファイルの場所を指定する「Scratch Directory」ポップアップには、以下の選択肢があります:
- Use GAUSS_SCRDIR:Gaussianのスクラッチディレクトリーを使用(この環境変数名で指定します)
- Use launch directory:GaussViewを起動したディレクトリーを使用
- Specify:スクラッチディレクトリーとして使用する場所を指定
Windows環境下でのファイル指定
Windows環境下では、「Preferences」の「Desktop」でファイルの関連性を設定します。GaussViewに関連づけるファイル拡張子を指定すれば、その拡張子のファイルを開く時に自動的にGaussViewが起動するようになります(図59)。ファイル拡張子の設定は、2つのパネルに分かれています。
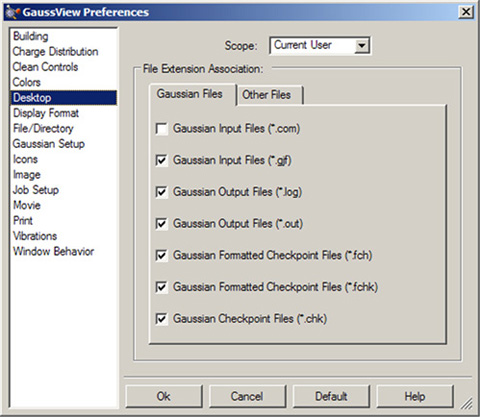
図59 Windowsのファイル関連性の指定
GaussViewとリストされたGaussianのファイルとの関連性を作成するには、対応するボックスにチェックを入れます。チェックを外すと、関連性は無くなります。「Scope」は、現在使用しているユーザーだけに設定が及ぶのかシステムの全ユーザーに及ぶのかを指定します。
|
